There are a couple of things I would just like to mention in passing. Jenny Ruhl has just posted a nice entry about ACCORD. This is very important. Lowering your blood glucose is significantly protective against CVD events. This is the exact opposite of the initial analysis of the results where a flawed interpretation of the data led to vociferous suggestions that lowering the HbA1c of diabetics might be actually dangerous. Hopefully this reanalysis will put an end to such stupid ideas which are still dangerously prevalent today.
Before Jenny posted the above I had been thinking about both metformin and insulin for the management of diabetes. I have posted on insulin, which is probably the ideal drug for diabetes management provided it is combined with low carbohydrate eating, in the past but it bears reiterating.
This is my opinion. If you can control your diabetes with metformin and wish to eat lots of carbohydrate, by all means get on with it, that's your choice. Not checking or not worrying about your blood glucose excursions might be a mistake. What you mean by good control might not involve HbA1c values in the 5% region or below.
If you need insulin to control your blood glucose, you have no choice. It's low carb. Live with it.
Why?
Some researchers (who write third rate papers using a rather inappropriate pancreatectomised dog model) are now starting to wake up to the fact that the pancreas secretes insulin in to the portal vein, not the subcutaneous tissues. In normal individuals insulin/glucose arrives at the liver via the portal vein and insulin facilitates transport of the glucose in to the liver, being metabolised in the process. Relatively little post pranial insulin or glucose penetrates to the peripheral circulation in a normal individual.
EDIT: Gretchen has kindly pointed out that the liver uptakes glucose through GLUT2, not GLUT4. The function of insulin, acting on its receptor (facilitating its degradation), is to suppress gluconeogenesis and hepatic glucose output. This is the correct function of the elevated portal insulin level. It makes no difference to the issues with peripheral vs portal insulin, but the correction is welcome. END EDIT
Once a person needs insulin to control their blood sugar levels they inject it subcutaneously. This will invariably elevate the systemic concentration of insulin. It will only modestly elevate the portal vein level. This is very important.
In the cited paper the dogs get a meal with 50% of calories from carbohydrate, 30% from fat and 20% from protein. In control dogs, instrumented but not pancreatectomised, the portal vein insulin after the meal is, at certain time points, ten times the peripheral systemic concentration. This is what is needed to allow the liver deal with the glucose load from the meal while simultaneously protecting the body from both hyperglycaemia and hyperinsulinaemia.
Subcutaneous insulin will make the rest of the body do the liver's job of clearing post prandial glucose, the liver can't manage because it never sees the requisite ten times the normal peripheral insulin concentration needed to deal effectively with the portal glucose load.
What happens when you use the rest of the body as a glucose sump? From the paper:
"Peripheral hyperinsulinemia is associated not only with increased risk of hypoglycemia, but also an increase in catechol and cortisol secretion and lipolysis [14], deleterious effects on vessel walls [6], ischemic heart disease, hypertension and hyperlipidemia [8], and abnormalities in hemostasis [10]"
As always, I would add that it's the obese, blind, legless person in the queue for dialysis who pays the bill for eating the carbohydrate.
This is the situation for all type I diabetics and the more advanced type 2 diabetics. The only route round it is to use intra peritoneal insulin which is, in part, absorbed through the mesenteric veins so is partially portal vein selective. There are, needless to say, a stack of complications to intra peritoneal insulin infusion. Tight control of glucose using subcutaneous insulin from a blood glucose controlled pump is no solution. Though glycaemia is better controlled it is still at the cost of too little insulin in the portal vein and too much in the periphery, using the body as a glucose sump. Over the years I have never been quite able to decide whether hyperinsulinaemia or hyperglycaemia is the primary factor which kills nerves and kidneys. It's a difficult call. And a fascinating discussion in its own right.
What happens if you eat a diet very low in starch?
Very little insulin is ever secreted by the pancreas, especially as glucokinase down regulates. Very little glucose ever needs to be taken up by the liver. Very little insulin will be metabolised by the liver. The insulin gradient between the portal vein and the systemic circulation will be as low as you can practically get it. If someone still needs to inject insulin alongside a very low carbohydrate diet, and many might not, injecting a very small amount subcutaneously will deliver an arterial concentration to the gut, pancreas and eventually to the portal vein and liver which is still quite close to what the portal vein might have supplied. If the body is not using insulin the tissues will not extract it, so portal and systemic concentrations will converge. Everything pans out at some near basal level.
A very low carbohydrate diet is not perfect for insulin dependent diabetics but it is streets ahead of anything else. What people do or do not consider a "normal" human diet will not get around this. Need exogenous insulin? You are not in a position to eat ancestral starch. It's a simple matter of anatomy, physiology and biochemistry.
Peter
Thursday, December 04, 2014
Thursday, November 27, 2014
The P479L gene for CPT-1a and fatty acid oxidation
In order to work out what is happening with a given child having an episode of hypoglycaemia as a result of having the P479L version of CPT-1a, we need some information.
My thanks to Mike Eades for the full text of the paper on the Canadian Inuit, which does include a certain amount of useful clinical data.
Here is the snippet about a young girl having a hypoglycaemic episode while hospitalised:
“Plasma free fatty acid was 3.8 mmol/L and plasma 3-hydroxybutyrate was 0.5 mmol/L”
Blood glucose was 1.9 mmol/l at the time. An FFA level of 3,800 micromol/l is impressively high. She was generating a small amount of ketones.
No one would argue with intravenous glucose at this point, the question is about how she got here.
So. The problem here does not (as I'd initially thought) appear to insulin induced suppression of FFAs to a level at which beta oxidation fails to support metabolism. FFAs are very high, even for an P479L person after a short fast. With ketones starting to be produced (and low blood glucose) I feel it is reasonable to assume that her liver glycogen is depleted and, while some fatty acids are entering the hepatocytes, not enough of them are being oxidised to support ketogenesis. Glycogen is being depleted to keep liver cells functional. Gluconeogenesis from protein is unable to meet the hepatic (and whole body) demand for glucose calories in the situation of limited access to FFA calories.
However much glycogen derived glucose you consider that the ancestral diet contained I feel it is very, very unlikely to be greater than the glucose and fructose of a modern diet. I feel that getting enough glycogen in to the liver to fully fuel its metabolism in the absence of adequate fatty acid oxidation is a non starter. The P479L mutation was not "permitted" by high oral carb loading, it was permitted by conditions which facilitated fatty acid oxidation. You don't have to agree.
What starts to look much more interesting is what controls CPT-1a activity and how this might vary from the ancestral diet to the modern diet.
The paper makes the point that omega 3 fatty acids appear to up regulate fatty acid oxidation (in rats at least) by the liver. If this is true in humans then a high level of omega 3 fatty acids from marine fats might up regulate fatty acid oxidation to a level which no longer necessitates the depletion of hepatic glycogen derived form oral glucose intake or protein catabolism.
In support of this is that the distribution of P479L within Alaska is not uniform, it's significantly commoner in the coastal regions compared to the inland areas.
"The allele frequency and rate of homozygosity for the CPT-1a P479L variant were high in Inuit and Inuvialuit who reside in northern coastal regions. The variant is present at a low frequency in First Nations populations, who reside in areas less coastal than the Inuit or Inuvialuit in the two western territories"
I'm open to other explanations, there are papers suggesting that the mutation helps to preferentially dispose of omega 6 PUFA, with omega 3 fatty acids as the facilitator.
In summary: Maintaining adequate FFA oxidation to avoid glycogen depletion looks to be the core need in P479L. A high fat diet with a large proportion of omega 3 fats might be a plausible way of maintaining adequate hepatic fatty acid oxidation. Hyperglycaemia (via Crabtree effect) looks to be anathema. Glycogen loading with a normal starch/sugar based modern diet is clearly ineffective to prevent hypoglycaemia for some individuals. Resistant starch as a reliable nightly adjunct to infant feeding seems very unlikely in the ancestral diet. Repeated periods of fasting were probably routine when hunting was poor and does not appear to have selected against P479L in weaned children. Unweaned children are unlikely to be exposed to fasting, provided milk was available from lactation.
Well, there are some more thoughts on the biochemistry.
People clearly have very differing ideas of what the Inuit did or did not eat as an ancestral diet. The P479L gene eliminates the need for source of dietary glucose to explain very limited levels of ketosis recorded in the Inuit. While it is perfectly possible to invoke a high protein diet to explain a lack of ketosis in the fed state this goes nowhere towards explaining the limited ketosis of fasting. P479L fits perfectly well as an explanation.
I have some level of discomfort with using the Inuit as poster people for a ketogenic diet. That's fine. They may well have eaten what would be a ketogenic diet for many of us, but they certainly did not develop high levels of ketones when they carried the P479L gene.
However. Over the months Wooo and I seem to have come to some sort of conclusion that, while systemic ketones are a useful adjunct, a ketogenic diet is essentially a fatty acid based diet with minimal glucose excursions and maximal beta oxidation. Exactly how important the ketones themselves are is not quite so clear cut. From the Hyperlipid and Protons perspective I would be looking to maximise input to the electron transport chain as FADH2 at electron-transferring-flavoprotein dehydrogenase and minimise NADH input at complex I. Ketones do not do this. Ketones input at complex II, much as beta oxidation inputs at ETFdh, but ketones also generate large amounts of NADH in the process of turning the TCA from acetyl-CoA to get to complex II, which ETFdh does not. I'm not a great lover of increasing the ratio of NADH to NAD+. These are my biases.
Confirming that the Inuit are not poster boys for ketosis is a "so what?" moment for me. Using their P479L mutation to argue against ketogenic diets is more of a problem. It's a massive dis-service to any one of the many, many people out there who are eating their way in to metabolic syndrome to suggest that a ketogenic diet is a Bad Thing because no one has lived in ketosis before. Even the Inuit didn't! My own feeling is that everyone comes from stock who occasionally practiced and survived intermittent fasting so we are should be adapted to this. I'd guess that if you are of Siberian, Inuit or First Nations extraction you might benefit from Jay Wortman's oolichan oil as part of a ketogenic diet.
I'm always amazed by the concept that a ketogenic diet might be temporarily therapeutic but must be discontinued because it eventually becomes Bad For You. It reminds me so much of the converse concept that low fat diets, which might worsen every marker of health which people may care to look at, will deliver major benefits at some mythical future date.
Ultimately, point scoring on the internet about what the Inuit did or didn't eat shouldn't destroy people's chances of health. Destroying a circular argument about Inuit diets may may the destructor feel good. Destroying the feet, eyes and kidneys of a person with type 2 diabetes, who need a ketogenic diet, as a spin off from that victory must be difficult to live with. I don't know how anyone can do this.
I think that's probably all I have to say for now.
Peter
My thanks to Mike Eades for the full text of the paper on the Canadian Inuit, which does include a certain amount of useful clinical data.
Here is the snippet about a young girl having a hypoglycaemic episode while hospitalised:
“Plasma free fatty acid was 3.8 mmol/L and plasma 3-hydroxybutyrate was 0.5 mmol/L”
Blood glucose was 1.9 mmol/l at the time. An FFA level of 3,800 micromol/l is impressively high. She was generating a small amount of ketones.
No one would argue with intravenous glucose at this point, the question is about how she got here.
So. The problem here does not (as I'd initially thought) appear to insulin induced suppression of FFAs to a level at which beta oxidation fails to support metabolism. FFAs are very high, even for an P479L person after a short fast. With ketones starting to be produced (and low blood glucose) I feel it is reasonable to assume that her liver glycogen is depleted and, while some fatty acids are entering the hepatocytes, not enough of them are being oxidised to support ketogenesis. Glycogen is being depleted to keep liver cells functional. Gluconeogenesis from protein is unable to meet the hepatic (and whole body) demand for glucose calories in the situation of limited access to FFA calories.
However much glycogen derived glucose you consider that the ancestral diet contained I feel it is very, very unlikely to be greater than the glucose and fructose of a modern diet. I feel that getting enough glycogen in to the liver to fully fuel its metabolism in the absence of adequate fatty acid oxidation is a non starter. The P479L mutation was not "permitted" by high oral carb loading, it was permitted by conditions which facilitated fatty acid oxidation. You don't have to agree.
What starts to look much more interesting is what controls CPT-1a activity and how this might vary from the ancestral diet to the modern diet.
The paper makes the point that omega 3 fatty acids appear to up regulate fatty acid oxidation (in rats at least) by the liver. If this is true in humans then a high level of omega 3 fatty acids from marine fats might up regulate fatty acid oxidation to a level which no longer necessitates the depletion of hepatic glycogen derived form oral glucose intake or protein catabolism.
In support of this is that the distribution of P479L within Alaska is not uniform, it's significantly commoner in the coastal regions compared to the inland areas.
"The allele frequency and rate of homozygosity for the CPT-1a P479L variant were high in Inuit and Inuvialuit who reside in northern coastal regions. The variant is present at a low frequency in First Nations populations, who reside in areas less coastal than the Inuit or Inuvialuit in the two western territories"
I'm open to other explanations, there are papers suggesting that the mutation helps to preferentially dispose of omega 6 PUFA, with omega 3 fatty acids as the facilitator.
In summary: Maintaining adequate FFA oxidation to avoid glycogen depletion looks to be the core need in P479L. A high fat diet with a large proportion of omega 3 fats might be a plausible way of maintaining adequate hepatic fatty acid oxidation. Hyperglycaemia (via Crabtree effect) looks to be anathema. Glycogen loading with a normal starch/sugar based modern diet is clearly ineffective to prevent hypoglycaemia for some individuals. Resistant starch as a reliable nightly adjunct to infant feeding seems very unlikely in the ancestral diet. Repeated periods of fasting were probably routine when hunting was poor and does not appear to have selected against P479L in weaned children. Unweaned children are unlikely to be exposed to fasting, provided milk was available from lactation.
Well, there are some more thoughts on the biochemistry.
People clearly have very differing ideas of what the Inuit did or did not eat as an ancestral diet. The P479L gene eliminates the need for source of dietary glucose to explain very limited levels of ketosis recorded in the Inuit. While it is perfectly possible to invoke a high protein diet to explain a lack of ketosis in the fed state this goes nowhere towards explaining the limited ketosis of fasting. P479L fits perfectly well as an explanation.
I have some level of discomfort with using the Inuit as poster people for a ketogenic diet. That's fine. They may well have eaten what would be a ketogenic diet for many of us, but they certainly did not develop high levels of ketones when they carried the P479L gene.
However. Over the months Wooo and I seem to have come to some sort of conclusion that, while systemic ketones are a useful adjunct, a ketogenic diet is essentially a fatty acid based diet with minimal glucose excursions and maximal beta oxidation. Exactly how important the ketones themselves are is not quite so clear cut. From the Hyperlipid and Protons perspective I would be looking to maximise input to the electron transport chain as FADH2 at electron-transferring-flavoprotein dehydrogenase and minimise NADH input at complex I. Ketones do not do this. Ketones input at complex II, much as beta oxidation inputs at ETFdh, but ketones also generate large amounts of NADH in the process of turning the TCA from acetyl-CoA to get to complex II, which ETFdh does not. I'm not a great lover of increasing the ratio of NADH to NAD+. These are my biases.
Confirming that the Inuit are not poster boys for ketosis is a "so what?" moment for me. Using their P479L mutation to argue against ketogenic diets is more of a problem. It's a massive dis-service to any one of the many, many people out there who are eating their way in to metabolic syndrome to suggest that a ketogenic diet is a Bad Thing because no one has lived in ketosis before. Even the Inuit didn't! My own feeling is that everyone comes from stock who occasionally practiced and survived intermittent fasting so we are should be adapted to this. I'd guess that if you are of Siberian, Inuit or First Nations extraction you might benefit from Jay Wortman's oolichan oil as part of a ketogenic diet.
I'm always amazed by the concept that a ketogenic diet might be temporarily therapeutic but must be discontinued because it eventually becomes Bad For You. It reminds me so much of the converse concept that low fat diets, which might worsen every marker of health which people may care to look at, will deliver major benefits at some mythical future date.
Ultimately, point scoring on the internet about what the Inuit did or didn't eat shouldn't destroy people's chances of health. Destroying a circular argument about Inuit diets may may the destructor feel good. Destroying the feet, eyes and kidneys of a person with type 2 diabetes, who need a ketogenic diet, as a spin off from that victory must be difficult to live with. I don't know how anyone can do this.
I think that's probably all I have to say for now.
Peter
Sunday, November 16, 2014
Coconuts and Cornstarch in the Arctic?
EDIT There is a follow on post to this one including some clinical data on the hypoglycaemia episodes. I'll put a link in here now it's up. END EDIT
Remi and Ken both pointed me toward this paper:
A Selective Sweep on a Deleterious Mutation in CPT1A in Arctic Populations
The paper itself is largely an account of the detective work involved in pinning down a specific mutation which has been positively selected for in a Siberian population living in the Arctic. The same mutation is also present in non related groups inhabiting the Arctic areas of northern America. The mutated gene is very common and frequently homozygous. It puts a leucine in the place of a proline in CPT-1a, the core enzyme for getting long chain fatty acids in to mitochondria. Putting a leucine where there should be a proline means the protein is basically f*cked. The mutation is linked, not surprisingly, to failure to generate ketones in infancy and can be associated with profound hypoglycaemia, potentially causing sudden death.
From the evolutionary point of view we have here a mutation which is significantly lethal at well below reproductive age, so it should have been weeded out because affected individuals are less likely to live long enough to pass on the gene. But it has been highly positively selected for in several populations, the common factors being cold climate and minimal access to dietary carbohydrate. It's a paradox.
Following a link in the paper gives us this abstract, with this snippet:
"Investigation of seven patients from three families suspected of a fatty acid oxidation defect showed mean CPT-I enzyme activity of 5.9 ± 4.9 percent of normal controls"
A value 6% with an SD of 5% suggests to me that some of these people may well have a CPT-1a function very close to zero. How common is the mutation?
"We screened 422 consecutive newborns from the region of one of the Inuit families for this variant; 294 were homozygous, 103 heterozygous, and only 25 homozygous normal; thus the frequency of this variant allele is 0.81"
I think "very common" is a reasonable description.
How dangerous is it?
"Three of the seven patients and two cousins had hypoketotic hypoglycemia attributable to CPT-Ia deficiency"
Quite dangerous.
The next thing we can do is google CPT-1a deficiency and have a look what needs to be done to stay alive if you carry this gene.
Clearly, if you can't transport LCFAs in to your mitochondria, you should run your metabolism on glucose/pyruvate and avoid the dysfunctional fatty acid transporter. This means raw corn starch, as we have seen used (probably wrongly) for glycogen storage diseases. Properly cooked starches are too short acting to reliably keep a child alive all through the night. They aren't safe enough.
Of course MCT oils have a role too. A CPT-1a defect has no effect on MCT metabolism so these can be used either directly by tissues or indirectly via liver/glial produced ketones.
LCFAs, unable to be metabolised, accumulate in the tissues as a storage disease. The advice is to avoid them as far as possible.
So the archetypical CPT-1a defect tolerant environment would seem to be a person sitting on a South Seas Island beach by a pile of coconuts chewing on a raw yam, with copious flatus night and day.
But it's not.
The CPT-1a defect evolved in multiple non related populations where both starch and MCT were very notable by their near-complete absence. It's an Arctic selected gene. No starches. No coconuts.
Let's take a speculative look at what is going on.
Living on a very low carbohydrate diet is associated with chronically elevated free fatty acids, chronically low levels of insulin and an ignorance of glucose. i.e. the body ignores glucose. Synthesise what glucose is needed but, beyond that, who cares?
Living in a sea of free fatty acids, which are taken up in to cells in a largely concentration dependent manner, allows an increased gradient to push FFA-CoA at any residual function in CPT-1a. It would appear, from the evolutionary perspective of Arctic inhabitants, that near ketogenic levels of FFAs are adequate even if you have the proline to leucine substitution at amino acid 479 in CPT-1a. You can do enough beta oxidation to cope.
Of course, the minute you lower free fatty acids, perhaps to the level of a post prandial starchivore, beta oxidation is going to grind to a halt without the concentration gradient effect. This is pathological. The temporary fix of substrate level ATP synthesis and related pyruvate supply to the mitochondria is fine for a while, but any reactive hypoglycaemia is going to be potentially fatal, especially if you are asleep or food deprived at the time. We know that insulin suppresses lipolysis at levels which don't budge GLUT4s. When insulin has suppressed lipolysis and blood glucose is low, FFAs might be fatally limited.
If you have the mutation but you never do the starchivore thing your FFAs are high 24/7, whether you have just chewed on a lump of seal blubber or not. No paper in the reference list appears to have looked at the FFA levels of children with this mutation on a mixed diet, let alone on the ancestral fat based diet of the polar regions. Given sustained very high levels of FFAs, you might even make some ketones.
If free fatty acids are high and there is no insulin to divert them in to storage, all of the nasty storage diseases associated with CPT-1a dysfunction might well disappear. This is the situation where the mutation allows carriers to thrive.
I think elevated free fatty acids, without elevated insulin, is a recipe for the tolerance of this mutation.
But the mutation is not just tolerated. This is no neutral mutation, it is positively advantageous. The prevalence of the mutated gene is far from random. Why is it beneficial?
This is not quite so simple.
Uncoupling is one component. Uncoupling respiration generates heat. There might just be a positive advantage to running your metabolism fairly uncoupled in a very low temperature environment. Elevated FFAs are completely essential to uncoupling and heat generation. Limiting fatty acid removal from the cytoplasm to the mitochondria might be a facilitator of uncoupling. It's FFAs on the cytosolic side of UCPs which facilitate proton translocation. Having a higher level of cytoplasmic FFAs at a given level of plasma FFAs might give an advantage over the normal level of uncoupling seen under near ketogenic diet conditions.
The second possibility is that, once you have established high enough levels of FFAs to push through the CPT-1a bottle neck, you simply run at this level flat out, all the time. One of the features of the CPT-1a from the modified gene is that it fails to be inhibited by malonyl-CoA. Even with limited CPT-1a activity there must be times at which ATP synthesis exceeds metabolic requirements and fatty acid transport ought to slow. There is no longer any brake to be applied to FFA transport if excess acetyl-CoA, exported to form malonyl-CoA in the cytoplasm, fails to inhibit CPT-1a . Oversupply of ATP within the matrix is likely to provide optimal uncoupling conditions, in excess of those from a ketogenic diet with regulated fatty acid uptake. That would be my guess. If it's cold enough, this might make the difference between survival or not. It keeps you warm, especially when you are asleep and the TCA should be quiescent.
Flicking through other references in the paper it does appear that indigenous Siberian people do have an elevated resting metabolic rate. In fat free mass it is 17% above calculated values i.e. they are uncoupled.
Finally, adults are not affected by the hypoglycaemia syndrome. My presumption is that, after puberty, they are sufficiently insulin resistant to have adequate FFAs present to maintain relatively normal mitochondrial function. It's the children who need their ancestral diet.
People with glycogen storage diseases die of hypoglycaemia (amongst other problems). We know that a deeply ketogenic diet both protects from hypoglycaemia and sets the body up to run perfectly well without any dietary glucose, which might be lost to glycogen stored permanently in the liver/muscles. There is every justification for giving the finger to cornstarch here and the folks suggesting a modification of ketogenic eating appear to be on fairly safe biochemical ground.
For the P497L mutation everything from the evolutionary perspective suggest that a very high FFA inducing diet may be equally efficacious. But the risks associated with failure, from the occasional safe starch meal or unsafe birthday cake at a party, carries the potential for catastrophe once insulin puts free fatty acids in to free fall.
Peter
BTW: You just have to wonder if any other CPT-1 mutations might behave in a similar manner to the P497L change in the Arctic... Could it be bye-bye time for cornstarch?
Remi and Ken both pointed me toward this paper:
A Selective Sweep on a Deleterious Mutation in CPT1A in Arctic Populations
The paper itself is largely an account of the detective work involved in pinning down a specific mutation which has been positively selected for in a Siberian population living in the Arctic. The same mutation is also present in non related groups inhabiting the Arctic areas of northern America. The mutated gene is very common and frequently homozygous. It puts a leucine in the place of a proline in CPT-1a, the core enzyme for getting long chain fatty acids in to mitochondria. Putting a leucine where there should be a proline means the protein is basically f*cked. The mutation is linked, not surprisingly, to failure to generate ketones in infancy and can be associated with profound hypoglycaemia, potentially causing sudden death.
From the evolutionary point of view we have here a mutation which is significantly lethal at well below reproductive age, so it should have been weeded out because affected individuals are less likely to live long enough to pass on the gene. But it has been highly positively selected for in several populations, the common factors being cold climate and minimal access to dietary carbohydrate. It's a paradox.
Following a link in the paper gives us this abstract, with this snippet:
"Investigation of seven patients from three families suspected of a fatty acid oxidation defect showed mean CPT-I enzyme activity of 5.9 ± 4.9 percent of normal controls"
A value 6% with an SD of 5% suggests to me that some of these people may well have a CPT-1a function very close to zero. How common is the mutation?
"We screened 422 consecutive newborns from the region of one of the Inuit families for this variant; 294 were homozygous, 103 heterozygous, and only 25 homozygous normal; thus the frequency of this variant allele is 0.81"
I think "very common" is a reasonable description.
How dangerous is it?
"Three of the seven patients and two cousins had hypoketotic hypoglycemia attributable to CPT-Ia deficiency"
Quite dangerous.
The next thing we can do is google CPT-1a deficiency and have a look what needs to be done to stay alive if you carry this gene.
Clearly, if you can't transport LCFAs in to your mitochondria, you should run your metabolism on glucose/pyruvate and avoid the dysfunctional fatty acid transporter. This means raw corn starch, as we have seen used (probably wrongly) for glycogen storage diseases. Properly cooked starches are too short acting to reliably keep a child alive all through the night. They aren't safe enough.
Of course MCT oils have a role too. A CPT-1a defect has no effect on MCT metabolism so these can be used either directly by tissues or indirectly via liver/glial produced ketones.
LCFAs, unable to be metabolised, accumulate in the tissues as a storage disease. The advice is to avoid them as far as possible.
So the archetypical CPT-1a defect tolerant environment would seem to be a person sitting on a South Seas Island beach by a pile of coconuts chewing on a raw yam, with copious flatus night and day.
But it's not.
The CPT-1a defect evolved in multiple non related populations where both starch and MCT were very notable by their near-complete absence. It's an Arctic selected gene. No starches. No coconuts.
Let's take a speculative look at what is going on.
Living on a very low carbohydrate diet is associated with chronically elevated free fatty acids, chronically low levels of insulin and an ignorance of glucose. i.e. the body ignores glucose. Synthesise what glucose is needed but, beyond that, who cares?
Living in a sea of free fatty acids, which are taken up in to cells in a largely concentration dependent manner, allows an increased gradient to push FFA-CoA at any residual function in CPT-1a. It would appear, from the evolutionary perspective of Arctic inhabitants, that near ketogenic levels of FFAs are adequate even if you have the proline to leucine substitution at amino acid 479 in CPT-1a. You can do enough beta oxidation to cope.
Of course, the minute you lower free fatty acids, perhaps to the level of a post prandial starchivore, beta oxidation is going to grind to a halt without the concentration gradient effect. This is pathological. The temporary fix of substrate level ATP synthesis and related pyruvate supply to the mitochondria is fine for a while, but any reactive hypoglycaemia is going to be potentially fatal, especially if you are asleep or food deprived at the time. We know that insulin suppresses lipolysis at levels which don't budge GLUT4s. When insulin has suppressed lipolysis and blood glucose is low, FFAs might be fatally limited.
If you have the mutation but you never do the starchivore thing your FFAs are high 24/7, whether you have just chewed on a lump of seal blubber or not. No paper in the reference list appears to have looked at the FFA levels of children with this mutation on a mixed diet, let alone on the ancestral fat based diet of the polar regions. Given sustained very high levels of FFAs, you might even make some ketones.
If free fatty acids are high and there is no insulin to divert them in to storage, all of the nasty storage diseases associated with CPT-1a dysfunction might well disappear. This is the situation where the mutation allows carriers to thrive.
I think elevated free fatty acids, without elevated insulin, is a recipe for the tolerance of this mutation.
But the mutation is not just tolerated. This is no neutral mutation, it is positively advantageous. The prevalence of the mutated gene is far from random. Why is it beneficial?
This is not quite so simple.
Uncoupling is one component. Uncoupling respiration generates heat. There might just be a positive advantage to running your metabolism fairly uncoupled in a very low temperature environment. Elevated FFAs are completely essential to uncoupling and heat generation. Limiting fatty acid removal from the cytoplasm to the mitochondria might be a facilitator of uncoupling. It's FFAs on the cytosolic side of UCPs which facilitate proton translocation. Having a higher level of cytoplasmic FFAs at a given level of plasma FFAs might give an advantage over the normal level of uncoupling seen under near ketogenic diet conditions.
The second possibility is that, once you have established high enough levels of FFAs to push through the CPT-1a bottle neck, you simply run at this level flat out, all the time. One of the features of the CPT-1a from the modified gene is that it fails to be inhibited by malonyl-CoA. Even with limited CPT-1a activity there must be times at which ATP synthesis exceeds metabolic requirements and fatty acid transport ought to slow. There is no longer any brake to be applied to FFA transport if excess acetyl-CoA, exported to form malonyl-CoA in the cytoplasm, fails to inhibit CPT-1a . Oversupply of ATP within the matrix is likely to provide optimal uncoupling conditions, in excess of those from a ketogenic diet with regulated fatty acid uptake. That would be my guess. If it's cold enough, this might make the difference between survival or not. It keeps you warm, especially when you are asleep and the TCA should be quiescent.
Flicking through other references in the paper it does appear that indigenous Siberian people do have an elevated resting metabolic rate. In fat free mass it is 17% above calculated values i.e. they are uncoupled.
Finally, adults are not affected by the hypoglycaemia syndrome. My presumption is that, after puberty, they are sufficiently insulin resistant to have adequate FFAs present to maintain relatively normal mitochondrial function. It's the children who need their ancestral diet.
People with glycogen storage diseases die of hypoglycaemia (amongst other problems). We know that a deeply ketogenic diet both protects from hypoglycaemia and sets the body up to run perfectly well without any dietary glucose, which might be lost to glycogen stored permanently in the liver/muscles. There is every justification for giving the finger to cornstarch here and the folks suggesting a modification of ketogenic eating appear to be on fairly safe biochemical ground.
For the P497L mutation everything from the evolutionary perspective suggest that a very high FFA inducing diet may be equally efficacious. But the risks associated with failure, from the occasional safe starch meal or unsafe birthday cake at a party, carries the potential for catastrophe once insulin puts free fatty acids in to free fall.
Peter
BTW: You just have to wonder if any other CPT-1 mutations might behave in a similar manner to the P497L change in the Arctic... Could it be bye-bye time for cornstarch?
Tuesday, October 28, 2014
Are ketone esters dangerous?
Back in 1995 Veech was looking at a ketone mixture as physiologically equivalent to insulin/glucose. In order to limit his variables the isolated rat myocardia used in the study were perfused with Krebs-Henseleit buffer containing the metabolic milieu of interest. The buffer has no free fatty acids so takes the provision of acetyl CoA from beta oxidation right out of the equation. It also eliminates any uncoupling from free fatty acids in the perfusate. It took me a while to twig that this was potentially a very long way from the situation under fasting or ketogenic diet conditions where free fatty acids might well be at the maximal physiological levels whenever ketones hit 5.0mmol/l.
The idea was certainly in mind when the group published this, in 2004:
“Current ketogenic diets are all characterized by elevations of free fatty acids, which may lead to metabolic inefficiency by activation of the PPAR system and its associated uncoupling mitochondrial uncoupling proteins. New diets comprised of ketone bodies themselves or their esters may obviate this present difficulty.”
By 2012 the problem with ketogenic diets had been reduced to one of impossible compliance, rather than metabolic inefficiency of free fatty acid metabolism:
"Further, to achieve effective ketosis with KG diets, almost complete avoidance of carbohydrates is required to keep blood insulin levels low to maintain adipose tissue lipolysis. Such high-fat, no-carbohydrate diets are unpalatable, leading to poor patient compliance."
You notice the uncoupling, previously a potential problem, is now in the title of the paper. Ketones in real life, even from ketone esters, work in a milieu of free fatty acids. If you flood the mitochondria with ATP-generating ketones, which generate no ATP in the cytoplasm, you just might expect to open that uncoupling pore and allow a few FFAs to translocate some protons, to limit over production of ATP within the mitochondria.
Currently, in 2014, the delectable savour-the-flavour of ketone esters allows this:
“…the ester can be taken as an oral supplement without changing the habitual diet.”
I watch this stuff with some degree of amazement. There is a suspicion that AD incidence is increasing rather faster than an ageing population would explain. The suggestion is that it has an environmental component. Now, many potential explanations are possible but I would like to think it is the saturophobic, cholesterophobic, fructophilic low fat based dietary advice from the American Heart Association which is the prime driver. Seems likely.
If AD (also known as type 3 diabetes) is a dietary disease, much as type 2 diabetes is largely a dietary disease, providing a crutch which will allow you to cling to the diet which got you in to AD in the first place strikes me as the biggest risk from ketone esters.
Excepting the stale urine/sweaty socks yummy aroma of course. Bring on the egg yolks fried in butter as an alternative, please.
A ketogenic diet features several things in addition to ketones. There is the chronic normoglycaemia which is anathema to the Crabtree effect. There is the physiological rock bottom basement insulin levels in a system where insulin signalling is f*cked. There are the elevated free fatty acids. These are the best.
Those free fatty acids are taken up by astroglial cells and used to generate in-situ ketone bodies. What sort of levels do they supply in vivo? That's an unknown (as far as I can tell), but I'm willing to bet that FFA supply under true ketogenic eating is both high and consistent, irrespective of fed/fasted state.
This is not quite the case if you are on the old MCT kick or mainlining sweaty socks while munching crapinabag.
Peter
A little background about Dr and Mr Newport and ketones which triggered this post off:
I have been unable to tease out, from Dr Newport's original article, that of Emily Deans or from the abstract of the case report above, quite what level of carbohydrate Mr Newport consumed in the original MCT phase, during the drug trial or while on ketone esters. I suspect it might have been more than a banana a day.
Oh, and another addendum. I, personally, clearly have issues with faking a ketogenic diet. This is true. But let me not decry ketones or their esters per se. If MCT oil or ketone esters get you out of bed and let you get dressed without needing assistance, that's great. They sure as hell knock spots off of anything which Big Pharma has to offer for AD management. The fact that I have yet to die as a direct result of eating less than one banana a day means that I hope never to need ketone esters. I feel a ketogenic diet should be high on the agenda for those with neurodegenerative diseases, with ketone esters or MCTs as a fall back. But then I would, wouldn't I...
The idea was certainly in mind when the group published this, in 2004:
“Current ketogenic diets are all characterized by elevations of free fatty acids, which may lead to metabolic inefficiency by activation of the PPAR system and its associated uncoupling mitochondrial uncoupling proteins. New diets comprised of ketone bodies themselves or their esters may obviate this present difficulty.”
By 2012 the problem with ketogenic diets had been reduced to one of impossible compliance, rather than metabolic inefficiency of free fatty acid metabolism:
"Further, to achieve effective ketosis with KG diets, almost complete avoidance of carbohydrates is required to keep blood insulin levels low to maintain adipose tissue lipolysis. Such high-fat, no-carbohydrate diets are unpalatable, leading to poor patient compliance."
You notice the uncoupling, previously a potential problem, is now in the title of the paper. Ketones in real life, even from ketone esters, work in a milieu of free fatty acids. If you flood the mitochondria with ATP-generating ketones, which generate no ATP in the cytoplasm, you just might expect to open that uncoupling pore and allow a few FFAs to translocate some protons, to limit over production of ATP within the mitochondria.
Currently, in 2014, the delectable savour-the-flavour of ketone esters allows this:
“…the ester can be taken as an oral supplement without changing the habitual diet.”
I watch this stuff with some degree of amazement. There is a suspicion that AD incidence is increasing rather faster than an ageing population would explain. The suggestion is that it has an environmental component. Now, many potential explanations are possible but I would like to think it is the saturophobic, cholesterophobic, fructophilic low fat based dietary advice from the American Heart Association which is the prime driver. Seems likely.
If AD (also known as type 3 diabetes) is a dietary disease, much as type 2 diabetes is largely a dietary disease, providing a crutch which will allow you to cling to the diet which got you in to AD in the first place strikes me as the biggest risk from ketone esters.
Excepting the stale urine/sweaty socks yummy aroma of course. Bring on the egg yolks fried in butter as an alternative, please.
A ketogenic diet features several things in addition to ketones. There is the chronic normoglycaemia which is anathema to the Crabtree effect. There is the physiological rock bottom basement insulin levels in a system where insulin signalling is f*cked. There are the elevated free fatty acids. These are the best.
Those free fatty acids are taken up by astroglial cells and used to generate in-situ ketone bodies. What sort of levels do they supply in vivo? That's an unknown (as far as I can tell), but I'm willing to bet that FFA supply under true ketogenic eating is both high and consistent, irrespective of fed/fasted state.
This is not quite the case if you are on the old MCT kick or mainlining sweaty socks while munching crapinabag.
Peter
A little background about Dr and Mr Newport and ketones which triggered this post off:
I have been unable to tease out, from Dr Newport's original article, that of Emily Deans or from the abstract of the case report above, quite what level of carbohydrate Mr Newport consumed in the original MCT phase, during the drug trial or while on ketone esters. I suspect it might have been more than a banana a day.
Oh, and another addendum. I, personally, clearly have issues with faking a ketogenic diet. This is true. But let me not decry ketones or their esters per se. If MCT oil or ketone esters get you out of bed and let you get dressed without needing assistance, that's great. They sure as hell knock spots off of anything which Big Pharma has to offer for AD management. The fact that I have yet to die as a direct result of eating less than one banana a day means that I hope never to need ketone esters. I feel a ketogenic diet should be high on the agenda for those with neurodegenerative diseases, with ketone esters or MCTs as a fall back. But then I would, wouldn't I...
Sunday, October 12, 2014
Where has the superoxide gone?
This is the first section of Fig 1 section C from the paper using dihydroethidium (DHE) to view in vivo superoxide production in control and diabetic kidneys, though not in the figure below.

It's a very important figure as it shows, very convincingly, that sudden onset hyperglycaemia has zero effect, none whatsoever, on superoxide production in their model of normal, non diabetic kidney tissue, that's the second column, identical to the first.
I have a lot of time for the failure to generate superoxide in diabetic kidneys, especially with pyruvate dehydrogenase complex down regulation limiting input to mitochondria from the end stage of glycolysis. But I have a concept that acute hyperglycaemia in normal, non Crabtree affected, tissues SHOULD generate superoxide, it should come from the respiratory chain and it should more particularly come from complex I in the region of the FAD moiety, preferably via the FeS cluster N1-a.
Now, if I had an in vivo tool for viewing superoxide generation, how would I do this? Well, I would use it in vivo. I would set up an iv glucose infusion, or perhaps a large intragastric glucose bolus, inject the DHE, wait a while, then look for superoxide/DHE derivative with my lovely optical scanner.
To keep the scrutineers happy I might have repeated the findings ex vivo, using the technique of paramagnetic detection of a superoxide/spin trap derivative, but the core finding, that superoxide generation on acute hyperglycaemia does NOT occur has to be shown in vivo. We already know it DOES occur ex vivo in multiple models, and the authors cite the studies to show this.
So, if hyperglycaemia triggers superoxide generation ex vivo in assorted non Crabtree adapted cells, why doesn't it do so in this study?
I don't know. There is a piece of core information which the scrutineers failed (miserably) to demand to be included in the study methods.
Figure 1C was not obtained in vivo. Column Ctrl was from a tissue homogenate of health kidney from non diabetic mice fed with pyruvate, malonate and ADP, subsequently flooded with 25mmol of glucose to produce the +HG column. That is not so bad. It's a model and it's clearly able to get GrantAid quality results.
But is it real?
Let's look at the equipment used. This is what they say:
"These studies were carried out in a MiniScope MS200 Benchtop EPR Spectrometer (Magnettech), which is designed to allow tight control of pO2 and temperature".
Why do they need tight control of pO2? You can obtain utterly rigid control control of pO2 by exposing your preparation to room air. Correct pressure to 760mmHG and pO2 is fixed at 21% of this.
To me the question is: What was the pO2 which failed to generate any superoxide when a mush of cytosol and mitochondria was exposed to 25mmol of glucose?
Was it 159.6mmHg, i.e. room air? Was it 40-50mmHg as other groups suspect mitochondria run at? Or was it 22mmHg?
This might matter. I got the 22mmHg value from the previous paper by the same authors which gave 3% oxygen as the likely conditions for normal mitochondrial function. This was a non referenced, throw away comment:
"Because the physiologic concentration of oxygen in mammals in vivo is less than 3% in most organs, we carried out a series of studies to determine whether ethidium or 2-hydroxyethidium was the specific oxidation product of DHE in vivo (i.e., in the intact animal, not cell culture/tissue slice) using several different validated animal models of increased or decreased superoxide".
Why it matters to me so much is that if an electron is thrown out of complex I due to hyperglycaemia triggered reverse electron flow through complex I, would it generate superoxide if the pO2 had been set to below physiological limits? Or if the guesstimate of 3% oxygen is correct and there is no superoxide generated, is there no reverse flow occurring? Or does the reverse flow occur, the electron is ejected, but it drops on to the surrounding protein structure rather than oxygen to be used as a distant signal via superoxide/H2O2/insulin receptor?
Using the in vivo technique would have told us exactly what was happening, at a true but non measured tissue pO2. I'm worried that the in vivo technique showed the anticipated (by me) hyperglycaemic superoxide and an ex vivo technique had to be developed and adjusted to maintain the fund generating core finding of no extra superoxide.
There was no reply to a simple polite email query as to the pO2 used.
Peter

It's a very important figure as it shows, very convincingly, that sudden onset hyperglycaemia has zero effect, none whatsoever, on superoxide production in their model of normal, non diabetic kidney tissue, that's the second column, identical to the first.
I have a lot of time for the failure to generate superoxide in diabetic kidneys, especially with pyruvate dehydrogenase complex down regulation limiting input to mitochondria from the end stage of glycolysis. But I have a concept that acute hyperglycaemia in normal, non Crabtree affected, tissues SHOULD generate superoxide, it should come from the respiratory chain and it should more particularly come from complex I in the region of the FAD moiety, preferably via the FeS cluster N1-a.
Now, if I had an in vivo tool for viewing superoxide generation, how would I do this? Well, I would use it in vivo. I would set up an iv glucose infusion, or perhaps a large intragastric glucose bolus, inject the DHE, wait a while, then look for superoxide/DHE derivative with my lovely optical scanner.
To keep the scrutineers happy I might have repeated the findings ex vivo, using the technique of paramagnetic detection of a superoxide/spin trap derivative, but the core finding, that superoxide generation on acute hyperglycaemia does NOT occur has to be shown in vivo. We already know it DOES occur ex vivo in multiple models, and the authors cite the studies to show this.
So, if hyperglycaemia triggers superoxide generation ex vivo in assorted non Crabtree adapted cells, why doesn't it do so in this study?
I don't know. There is a piece of core information which the scrutineers failed (miserably) to demand to be included in the study methods.
Figure 1C was not obtained in vivo. Column Ctrl was from a tissue homogenate of health kidney from non diabetic mice fed with pyruvate, malonate and ADP, subsequently flooded with 25mmol of glucose to produce the +HG column. That is not so bad. It's a model and it's clearly able to get GrantAid quality results.
But is it real?
Let's look at the equipment used. This is what they say:
"These studies were carried out in a MiniScope MS200 Benchtop EPR Spectrometer (Magnettech), which is designed to allow tight control of pO2 and temperature".
Why do they need tight control of pO2? You can obtain utterly rigid control control of pO2 by exposing your preparation to room air. Correct pressure to 760mmHG and pO2 is fixed at 21% of this.
To me the question is: What was the pO2 which failed to generate any superoxide when a mush of cytosol and mitochondria was exposed to 25mmol of glucose?
Was it 159.6mmHg, i.e. room air? Was it 40-50mmHg as other groups suspect mitochondria run at? Or was it 22mmHg?
This might matter. I got the 22mmHg value from the previous paper by the same authors which gave 3% oxygen as the likely conditions for normal mitochondrial function. This was a non referenced, throw away comment:
"Because the physiologic concentration of oxygen in mammals in vivo is less than 3% in most organs, we carried out a series of studies to determine whether ethidium or 2-hydroxyethidium was the specific oxidation product of DHE in vivo (i.e., in the intact animal, not cell culture/tissue slice) using several different validated animal models of increased or decreased superoxide".
Why it matters to me so much is that if an electron is thrown out of complex I due to hyperglycaemia triggered reverse electron flow through complex I, would it generate superoxide if the pO2 had been set to below physiological limits? Or if the guesstimate of 3% oxygen is correct and there is no superoxide generated, is there no reverse flow occurring? Or does the reverse flow occur, the electron is ejected, but it drops on to the surrounding protein structure rather than oxygen to be used as a distant signal via superoxide/H2O2/insulin receptor?
Using the in vivo technique would have told us exactly what was happening, at a true but non measured tissue pO2. I'm worried that the in vivo technique showed the anticipated (by me) hyperglycaemic superoxide and an ex vivo technique had to be developed and adjusted to maintain the fund generating core finding of no extra superoxide.
There was no reply to a simple polite email query as to the pO2 used.
Peter
Friday, October 10, 2014
The Crabtree Effect and superoxide in diabetes
I started with this paper about in vivo superoxide detection in the brain but, apart from the technique, there was no examination of the response to hyperglycaemia so I moved on. The next paper by the same group is looking at superoxide and mitochondrial function/health in the kidney under various models of diabetes. The general principles appear similar in neurons and kidney cells.
An in vivo technique to view superoxide is really useful. I have alluded to a certain discomfort in examining electron/oxygen interaction in mitochondria within cells/mitochondrial preparations under room air, with a partial pressure for oxygen of around 150mmHg (sorry for the non SI units, showing my age there!). There is no way that normal mitochondria are exposed to this much oxygen, a little browse around pubmed suggests that the best in vivo estimate is around 40-50mmHg, subject to some debate. That's without even thinking about what CO2 partial pressure you should use for cell culture... So observation in vivo takes care of a lot of this. If an electron is thrown out of the respiratory chain (I feel nothing in the ETC is accidental) the chances of it dropping on to an oxygen molecule seem somewhat higher if we have three times the oxygen partial pressure than the system was designed to work under. If the electron wasn't destined for an oxygen molecule, where else might it have been going?
The first point has to be that in two models of type one diabetes there is less superoxide production in the kidneys of diabetic mice in vivo than control mice, that's Figure 1 section C. Going ex vivo (I probably have a full post on the problems with this ex vivo section) we have the same effect demonstrated using a paramagnetic technique, that's Fig 2C. The reduced superoxide in diabetic kidneys was confirmed in the tissue homogenates under relatively normal metabolic substrate supply. Exposing the preparations to glucose at 25mmol/l has no effect on superoxide generation from the control kidney homogenate but actually reduces it, rather a lot, in the diabetic derived homogenate.
Interesting.
NOTE If you follow the text through about Fig 1C and their SOD2+/- mice you will find that the data is not very accurately described. So caution here. The SOD2+/- had a non significant increase in mitochondrial superoxide in Fig 1C, so it is hardly surprising this did not rescue the diabetic mice from renal disease in Fig 3A and F. I don't like their writing about this whole SOD2+/- section. Definite caution. END NOTE.
The paper has, amongst its problems, a lot of very perceptive points which make a great deal of sense. It's quite hard to know where to start. Let's begin with the failure of superoxide production.
So this paper flies in the face of the Protons concept of hyperglycaemia driving reverse electron flow from mtG3Pdh through complex I to generate insulin resistance. That too is probably another post, comparing the diabetic state with the non diabetic hyperglycaemic state. Anyhoo. The group rather like Crabtree. So do I. The Crabtree effect, the shutting down/mothballing of mitochondrial function, is an adaptation to oversupply of glycolysis derived substrates. It allows a limit to be set on the throughput of pyruvate to mitochondria and jettisons any excess as lactate. This situation, once it is established, is probably quite different to the situation which leads to its adoption.
Chronic hyperglycaemia induces the Crabtree effect and down regulates mitochondrial biogenesis, mitochondrial repair and electron transport chain function. It not only does this but it also phosphorylates the pyruvate dehydrogenase complex, very specifically, and this directly shuts down input to the TCA from glycolysis (or input from lactate itself, if we want to apply this concept to neurons, as we might). This is all in the paper. Of course I would add that it doesn't affect ketone derived acetyl-CoA input to the TCA, although the ketone derived acetyl-CoA will be processed by a degenerate electron transport chain...
Under sustained hyperglycaemia there is an excess of calories which leads to a failure to activate AMP kinase, a core sensor of energy abundance which is phosphorylated under hypo caloric conditions. AMPK regulates PGC 1 alpha, a messenger to trigger mitochondrial biogenesis. But the central link, the activation of AMKP, is mitochondrial derived superoxide. And, oddly enough, one of the functions of AMPK activation is the generation of mitochondrial superoxide. A self sustaining loop.
The group administered rotenone to control mice. Now, the effect of rotenone on superoxide generation appears (in general) to be rather dose rate related. In the present study the dose rate was chosen so that there was a near complete suppression of superoxide production from the ETC of the mice. Acute suppression of superoxide results in the reduced phosphorylation (reduced activation) of AMPK and increased phosphorylation of PDH, which shuts it down. This loss of superoxide is a short term mimic of the long term established Crabtree effect. No superoxide, no mitochondrial maintenance. Consider that chronic high dose rotenone poisoning is a standard model for Parkinsons Disease and you begin to see the importance of superoxide in the brain. Long term hyperglycaemic failure to generate superoxide is probably a more normal route to neurodegeneration than rotenone in most (but not all) neurodegenerate humans...
The fall in superoxide production in diabetic tissue homogenates again pulls me back to brain function. Crabtree suppresses hyperglycaemic superoxide production, i.e. the effect is antioxidant. Let's look at what glucose does to neurons from this paper which we've chatted about before. Here's the only bit I'm interested in today:
"Indeed, it has been shown that glucose is used by neurons to maintain their antioxidant status via the pentose phosphate pathway (PPP), which cannot be fueled by lactate (Magistretti, 2008; Herrero-Mendez et al., 2009)"
It's impossible over emphasise the importance of that sentence. It says it all about why neurons should run on lactate! To avoid upregulating antioxidant status.
What does increasing antioxidant status do to superoxide signalling? The term f*cked comes (unavoidably South Park-ishly) to mind. There are a swathe of papers showing that the antioxidant status in neurons of AD and PD patients is upregulated.
Once you go with Crabtree you can see that glucose and PPP driven antioxidant upregulation might be all that is needed to lose superoxide signalling and destroy mitochondrial function. Lactate does not do this. Lactate does not induce the Crabtree effect.
Let's be very specific: Glucose, under the Crabtree effect, triggers a cascade which ends up with failure to generate superoxide and this maintains mitochondrial shutdown. Up regulating antioxidant status may theoretically be helpful in dealing with non mitochondrial superoxide generation, but it's not going to help signal for mitochondrial biogenesis.
High glucose exposure generates glucose dependence. This is a recurring theme and is core to neurodegeneration. I look at safe starches and can see that, if you are living with the Crabtree effect in key neurons, some starch/glycolysis might make you feel better if you are ketogenically hypoglycaemic, but it's not going to help un-Crabtree your mitochondria. On the other hand I can't see that pushing starch to a level which produces hyperglycaemia is anything other than damaging, as opposed to merely neutral as it might be when your pancreas does its job effectively.
I'll take a break before going on to the sections on mitochondrial deletions and respiratory chain oxidative damage elsewhere in the paper. Or maybe I should talk about the bits I deeply dislike related to oxygen pressure and superoxide.
Peter
An in vivo technique to view superoxide is really useful. I have alluded to a certain discomfort in examining electron/oxygen interaction in mitochondria within cells/mitochondrial preparations under room air, with a partial pressure for oxygen of around 150mmHg (sorry for the non SI units, showing my age there!). There is no way that normal mitochondria are exposed to this much oxygen, a little browse around pubmed suggests that the best in vivo estimate is around 40-50mmHg, subject to some debate. That's without even thinking about what CO2 partial pressure you should use for cell culture... So observation in vivo takes care of a lot of this. If an electron is thrown out of the respiratory chain (I feel nothing in the ETC is accidental) the chances of it dropping on to an oxygen molecule seem somewhat higher if we have three times the oxygen partial pressure than the system was designed to work under. If the electron wasn't destined for an oxygen molecule, where else might it have been going?
The first point has to be that in two models of type one diabetes there is less superoxide production in the kidneys of diabetic mice in vivo than control mice, that's Figure 1 section C. Going ex vivo (I probably have a full post on the problems with this ex vivo section) we have the same effect demonstrated using a paramagnetic technique, that's Fig 2C. The reduced superoxide in diabetic kidneys was confirmed in the tissue homogenates under relatively normal metabolic substrate supply. Exposing the preparations to glucose at 25mmol/l has no effect on superoxide generation from the control kidney homogenate but actually reduces it, rather a lot, in the diabetic derived homogenate.
Interesting.
NOTE If you follow the text through about Fig 1C and their SOD2+/- mice you will find that the data is not very accurately described. So caution here. The SOD2+/- had a non significant increase in mitochondrial superoxide in Fig 1C, so it is hardly surprising this did not rescue the diabetic mice from renal disease in Fig 3A and F. I don't like their writing about this whole SOD2+/- section. Definite caution. END NOTE.
The paper has, amongst its problems, a lot of very perceptive points which make a great deal of sense. It's quite hard to know where to start. Let's begin with the failure of superoxide production.
So this paper flies in the face of the Protons concept of hyperglycaemia driving reverse electron flow from mtG3Pdh through complex I to generate insulin resistance. That too is probably another post, comparing the diabetic state with the non diabetic hyperglycaemic state. Anyhoo. The group rather like Crabtree. So do I. The Crabtree effect, the shutting down/mothballing of mitochondrial function, is an adaptation to oversupply of glycolysis derived substrates. It allows a limit to be set on the throughput of pyruvate to mitochondria and jettisons any excess as lactate. This situation, once it is established, is probably quite different to the situation which leads to its adoption.
Chronic hyperglycaemia induces the Crabtree effect and down regulates mitochondrial biogenesis, mitochondrial repair and electron transport chain function. It not only does this but it also phosphorylates the pyruvate dehydrogenase complex, very specifically, and this directly shuts down input to the TCA from glycolysis (or input from lactate itself, if we want to apply this concept to neurons, as we might). This is all in the paper. Of course I would add that it doesn't affect ketone derived acetyl-CoA input to the TCA, although the ketone derived acetyl-CoA will be processed by a degenerate electron transport chain...
Under sustained hyperglycaemia there is an excess of calories which leads to a failure to activate AMP kinase, a core sensor of energy abundance which is phosphorylated under hypo caloric conditions. AMPK regulates PGC 1 alpha, a messenger to trigger mitochondrial biogenesis. But the central link, the activation of AMKP, is mitochondrial derived superoxide. And, oddly enough, one of the functions of AMPK activation is the generation of mitochondrial superoxide. A self sustaining loop.
The group administered rotenone to control mice. Now, the effect of rotenone on superoxide generation appears (in general) to be rather dose rate related. In the present study the dose rate was chosen so that there was a near complete suppression of superoxide production from the ETC of the mice. Acute suppression of superoxide results in the reduced phosphorylation (reduced activation) of AMPK and increased phosphorylation of PDH, which shuts it down. This loss of superoxide is a short term mimic of the long term established Crabtree effect. No superoxide, no mitochondrial maintenance. Consider that chronic high dose rotenone poisoning is a standard model for Parkinsons Disease and you begin to see the importance of superoxide in the brain. Long term hyperglycaemic failure to generate superoxide is probably a more normal route to neurodegeneration than rotenone in most (but not all) neurodegenerate humans...
The fall in superoxide production in diabetic tissue homogenates again pulls me back to brain function. Crabtree suppresses hyperglycaemic superoxide production, i.e. the effect is antioxidant. Let's look at what glucose does to neurons from this paper which we've chatted about before. Here's the only bit I'm interested in today:
"Indeed, it has been shown that glucose is used by neurons to maintain their antioxidant status via the pentose phosphate pathway (PPP), which cannot be fueled by lactate (Magistretti, 2008; Herrero-Mendez et al., 2009)"
It's impossible over emphasise the importance of that sentence. It says it all about why neurons should run on lactate! To avoid upregulating antioxidant status.
What does increasing antioxidant status do to superoxide signalling? The term f*cked comes (unavoidably South Park-ishly) to mind. There are a swathe of papers showing that the antioxidant status in neurons of AD and PD patients is upregulated.
Once you go with Crabtree you can see that glucose and PPP driven antioxidant upregulation might be all that is needed to lose superoxide signalling and destroy mitochondrial function. Lactate does not do this. Lactate does not induce the Crabtree effect.
Let's be very specific: Glucose, under the Crabtree effect, triggers a cascade which ends up with failure to generate superoxide and this maintains mitochondrial shutdown. Up regulating antioxidant status may theoretically be helpful in dealing with non mitochondrial superoxide generation, but it's not going to help signal for mitochondrial biogenesis.
High glucose exposure generates glucose dependence. This is a recurring theme and is core to neurodegeneration. I look at safe starches and can see that, if you are living with the Crabtree effect in key neurons, some starch/glycolysis might make you feel better if you are ketogenically hypoglycaemic, but it's not going to help un-Crabtree your mitochondria. On the other hand I can't see that pushing starch to a level which produces hyperglycaemia is anything other than damaging, as opposed to merely neutral as it might be when your pancreas does its job effectively.
I'll take a break before going on to the sections on mitochondrial deletions and respiratory chain oxidative damage elsewhere in the paper. Or maybe I should talk about the bits I deeply dislike related to oxygen pressure and superoxide.
Peter
Thursday, September 25, 2014
Uncoupling control in defence of FFAs
I've been reading this review on beta hydroxybutyrate and am struck by the concerns expressed throughout about the potential damage caused by free fatty acids, due to uncoupling, a sentiment I have picked up in several of Veech's publications which are heavily cited in the review.
I was particularly struck by how two papers I've recently discussed were described, so it's topical for me. One was the puzzling toxicity of a LCKD diet as published by Wang et al. This is the one using vegetable shortening of indeterminate trans fat concentration, a point sadly un-noted (or considered unimportant?) by the review. And second is the Kuwait study, described as LCKD in the review, which was not exactly glycogen depleting for a rodent.
Aside: This cited study starved rats for three days before ischaemia/reperfusion. That should have depleted glycogen AND raised raised FFAs (neither of which was checked, but any lipophobe should expect uncoupling combined with backup anaerobic glycogen reserve loss to be disastrous in ischaemia/reperfusion) as well as predictably increasing B-OHB. Combined starvation changes in fact reduce the damage produced and improve recovery. End aside.
So I'm a little ambivalent about the review and how much of the rest of their ideas I might take at face value.
Ultimately, thinking about free fatty acids, we have to talk about the control of uncoupling.
Recall this image from this study in part 29 of the Protons thread:

Free fatty acids are essential for proton transport across the inner mitochondrial membrane to uncouple oxygen consumption from ATP synthesis and to maximise electron flow down the electron transport chain with minimal resistance and minimal non essential superoxide generation.
No free fatty acids, no uncoupling. Free fatty acids are core to uncoupling.
But they are far from the only factor. For protons to be transported through the channel of the UCP by free fatty acids the channel must undergo a conformational change, which is highly dependent on the ATP status of the cytoplasm and the mitochondrial matrix.
So we have this picture from this very impressive study:

ATP in the cytoplasm fits in to a specific binding site, with each phosphate moiety of ATP fitting up against a specific arginine, all three aligning results in closure of the channel and inhibition of uncoupling, whatever the FFA concentration. Here is what the authors say:
"Moreover, residues R79 and R279 correspond to the arginines involved in nucleotide binding and protein inhibition in UCP1. According to the three-step binding model proposed for UCP1,17 β-phosphate of PN [phospho-nucleotide] binds first to R182 (helix IV, loose binding). The second step is the binding of γ-phosphate to R83 after protonation of E190 (tight binding). After the subsequent binding of α-phosphate to R276 (helix VI) the protein switches to the inhibited conformation"
Cytoplasmic ATP (and GTP) inhibit uncoupling. But not all of the time, despite the fact that there is normally always enough cytoplasmic ATP to inhibit uncoupling. So yet another factor comes in to play.
It is quite possible to inhibit the inhibition of uncoupling produced by cytoplasmic ATP.
You do this with mitochondrial ATP. ATP binding from the mitochondrial side of the channel interferes with the binding of cytoplasmic ATP but cannot reach the R83 arginine itself to close the channel. So elevated mitochondrial ATP keeps the uncoupling channel open, even in the face of rather high cytoplasmic ATP levels.
The logic to this is that if there is plenty of ATP within the mitochondria there is no need to preserve delta psi and it's fine to uncouple. If there is ATP in the cytoplasm but very little in the mitochondria the implication appears to be that ATP synthase is not generating enough mitochondrial ATP, i.e. we are either hypoxic or over-uncoupled. Continued glycolysis generates ATP on the cytoplasmic side so allows the uncoupling channel to close using this cytoplasmic ATP.
It's pretty logical.
So. Under hypoxia, whatever the level of FFAs, what happens to uncoupling?
It stops due to a lack of mitochondrial ATP. Should you fear FFAs? Only if you think you will continue to uncouple respiration under hypoxia. The balance of mitochondrial to cytoplasmic ATP should shut down uncoupling very rapidly when needed.
Just say no to Crisco (if that's how Wang et al got their result).
It has long worried me that in Veech's seminal paper on glucose, insulin and ketone metabolism in an isolated heart preparation the group was very, very careful to run the study without any involvement of free fatty acids. For those of us living in a temperate latitudes, lounging on the beach under a coconut palm while waiting for lunch to drop on our heads is not an option. Have you ever been to Lowestoft beach? No ketones without elevated FFAs at latitude 52 deg N on the North Sea coast. Fasting, or living on meat for a while, seems more likely than eating MCTs outside the tropics. I fail to see how the body would manufacture the miracle of ketones at exactly the same time as it releases the devil incarnate of free fatty acids.
Some folks like free fatty acids. Me, for one.
Some of us like uncoupling too, in the right place, at the right time.
Peter
I was particularly struck by how two papers I've recently discussed were described, so it's topical for me. One was the puzzling toxicity of a LCKD diet as published by Wang et al. This is the one using vegetable shortening of indeterminate trans fat concentration, a point sadly un-noted (or considered unimportant?) by the review. And second is the Kuwait study, described as LCKD in the review, which was not exactly glycogen depleting for a rodent.
Aside: This cited study starved rats for three days before ischaemia/reperfusion. That should have depleted glycogen AND raised raised FFAs (neither of which was checked, but any lipophobe should expect uncoupling combined with backup anaerobic glycogen reserve loss to be disastrous in ischaemia/reperfusion) as well as predictably increasing B-OHB. Combined starvation changes in fact reduce the damage produced and improve recovery. End aside.
So I'm a little ambivalent about the review and how much of the rest of their ideas I might take at face value.
Ultimately, thinking about free fatty acids, we have to talk about the control of uncoupling.
Recall this image from this study in part 29 of the Protons thread:

Free fatty acids are essential for proton transport across the inner mitochondrial membrane to uncouple oxygen consumption from ATP synthesis and to maximise electron flow down the electron transport chain with minimal resistance and minimal non essential superoxide generation.
No free fatty acids, no uncoupling. Free fatty acids are core to uncoupling.
But they are far from the only factor. For protons to be transported through the channel of the UCP by free fatty acids the channel must undergo a conformational change, which is highly dependent on the ATP status of the cytoplasm and the mitochondrial matrix.
So we have this picture from this very impressive study:

ATP in the cytoplasm fits in to a specific binding site, with each phosphate moiety of ATP fitting up against a specific arginine, all three aligning results in closure of the channel and inhibition of uncoupling, whatever the FFA concentration. Here is what the authors say:
"Moreover, residues R79 and R279 correspond to the arginines involved in nucleotide binding and protein inhibition in UCP1. According to the three-step binding model proposed for UCP1,17 β-phosphate of PN [phospho-nucleotide] binds first to R182 (helix IV, loose binding). The second step is the binding of γ-phosphate to R83 after protonation of E190 (tight binding). After the subsequent binding of α-phosphate to R276 (helix VI) the protein switches to the inhibited conformation"
Cytoplasmic ATP (and GTP) inhibit uncoupling. But not all of the time, despite the fact that there is normally always enough cytoplasmic ATP to inhibit uncoupling. So yet another factor comes in to play.
It is quite possible to inhibit the inhibition of uncoupling produced by cytoplasmic ATP.
You do this with mitochondrial ATP. ATP binding from the mitochondrial side of the channel interferes with the binding of cytoplasmic ATP but cannot reach the R83 arginine itself to close the channel. So elevated mitochondrial ATP keeps the uncoupling channel open, even in the face of rather high cytoplasmic ATP levels.
The logic to this is that if there is plenty of ATP within the mitochondria there is no need to preserve delta psi and it's fine to uncouple. If there is ATP in the cytoplasm but very little in the mitochondria the implication appears to be that ATP synthase is not generating enough mitochondrial ATP, i.e. we are either hypoxic or over-uncoupled. Continued glycolysis generates ATP on the cytoplasmic side so allows the uncoupling channel to close using this cytoplasmic ATP.
It's pretty logical.
So. Under hypoxia, whatever the level of FFAs, what happens to uncoupling?
It stops due to a lack of mitochondrial ATP. Should you fear FFAs? Only if you think you will continue to uncouple respiration under hypoxia. The balance of mitochondrial to cytoplasmic ATP should shut down uncoupling very rapidly when needed.
Just say no to Crisco (if that's how Wang et al got their result).
It has long worried me that in Veech's seminal paper on glucose, insulin and ketone metabolism in an isolated heart preparation the group was very, very careful to run the study without any involvement of free fatty acids. For those of us living in a temperate latitudes, lounging on the beach under a coconut palm while waiting for lunch to drop on our heads is not an option. Have you ever been to Lowestoft beach? No ketones without elevated FFAs at latitude 52 deg N on the North Sea coast. Fasting, or living on meat for a while, seems more likely than eating MCTs outside the tropics. I fail to see how the body would manufacture the miracle of ketones at exactly the same time as it releases the devil incarnate of free fatty acids.
Some folks like free fatty acids. Me, for one.
Some of us like uncoupling too, in the right place, at the right time.
Peter
Tuesday, September 23, 2014
Ketones for ALS?
OK. I've been thinking a lot about ketogenic diets and motor neuron disease, which appears to me to be just one facet of Alzheimers, Parkinsons and a number of other neurodegenerative diseases.
The first thing to say is that they (ketones) don't seem to work terribly well. I picked up this paper via the Deanna Protocol website. I wrote an unpublished post at the time setting down what an abysmally written paper it is but I thought I would stick to the basics today. Feeding a ketogenic diet to the mice engineered to have an ALS-like disease delays their time to falling off a log but does not extend their lifespan:
"There was no statistically significant difference in the age at death between KD fed animals compared to SOD1-G93 transgenic mice fed a standard laboratory diet (133 ± 4 vs. 131 ± 4 days, p = 0.914)"
Some improved motor function, for a while, may be worth having if you suffer from ALS but I don't think it's exactly a cure or remission.
Although the methods section is very reticent about the diet, it is high in carbohydrate (20% of calories) and protein (20% of calories), so must be MCT based to achieve ketosis.
The findings are confirmed by a nicely written, very clear paper using caprylic acid as a supplement to standard CIAB mouse chow:
"SOD1-G93A animals on caprylic triglyceride diet had a median survival of 135 days. Although it was longer than the median survival of SOD1-G93A animals on control diet (129 days), it did not reach statistical significance (Mantel-Cox test, p=0.165)"
Things weren't much better here in the Deanna Protocol paper. Their ketogenic diet supplied 77% of calories from fat, type unspecified, and essentially zero from carbohydrate. Protein was high at 22% of calories. This is what you get as a result:
"Although the mean survival of SOD1-G93A animals was longer in all three treatment groups than the control group, this difference reached statistical significance only in the KD+DP (4.2%, p = 0.006) and SD+DP groups (7.5%, p = 0.001, Fig. 5, Table 5, Data S2)."
i.e. the ketogenic diet, without the alpha ketoglutarate of the Deanna Protocol, was no better than control mice on CIAB.
All of which is quite interesting and should be quite depressing for groups working with MCTs, ketone esters or ketone salts as managements for neurodegenerative diseases.
We can say certain things about the first two studies. Using MCTs on a moderate to high carbohydrate diet is unlikely to lead to the metabolic changes of a true ketogenic diet. Normoglycaemia is probably not on the menu. It will not lead to the sort of effects of minimal carbohydrate, just adequate protein, very high fat diet. The effect of such a diet has been described as unique.
Of course, a few grams of MCTs on a diet of standard lab chow will generate ketones. That is hardly equivalent to a true ketogenic diet with its reduced glycaemia and basement value insulin levels.
As the paper on the Deanna protocol reports:
"Blood glucose was not significantly different between the diet groups", not exactly what was reported for mice eating D12336.
Ultimately, no one yet appears to have looked at a true ketogenic diet in ALS.
The focus is on the ketones. Ketones are good, but they are not magic. There are people who believe that the ketones themselves are simply a surrogate for very low insulin levels, which is magic (You know who you are Wooo!) and that the benefits of ketogenic diets may stem from the low insulin levels rather than the ketones per se, certainly for obesity management. For neurodegenration I find this idea very appealing. I think that the low glucose/insulin might be particularly important within the brain. I can't see that the work has been done yet, too much of a focus on ketones.
As something of an aside, the Deanna Protocol is interesting in its own right. The core supplement is (arginine-linked) alpha ketoglutarate. From the Protons point of view, if the alpha ketoglutarate enters the TCA at alpha ketoglutarate dehydrogenase and leaves it at malate, it would appear to be a very FADH2 selective input at complex II, generating an NADH:FADH2 ratio of 1:1, i.e. it is functioning as a rather specific FADH2 input. We're all aware that complex I dysfunction is a hallmark of neurodegenerative diseases and, in the absence of beta oxidation (we're in neurons here), complex II is the primary route in to the CoQ couple for electrons via FADH2. Along with mtG3Pdh of course, if that happens to be active. I can see the logic to using this AKG to push complex II without the excess rise in non-usable NADH, which large amounts of acetyl-CoA provide. I'm not surprised AKG is the core component of the Deanna Protocol and hats off to her father for picking this up.
A further aside, Deanna tried coconut oil, caprylic acid and MCTs early on in her disease. Not a lot of help. Adding extra acetyl-CoA from ketones will be of limited help in a condition with complex I dysfunction. My interest still lies in ketones combined with low blood glucose, not as an add on to healthy starches.
It is quite clear from the last post featuring cardiac ischaemia and ketones that any old ketones will do when hypoxia is the problem: Bring on the MCTs. Logically ketones are fats, part pre-oxidised in the liver, so require less oxygen to complete their metabolism in the cardiac muscle. They do not uncouple protons from oxidative phosphorylation either, which we will probably come back to. And while normal fatty acids do uncouple ox phos, this effect is (under normal circumstances) completely lost when mitochondrial ATP levels fall. This probably happens rather quickly under hypoxia.
The energetic failure of neurodegenerative diseases is only partially amenable to ketones. We are looking at a rather different phenomenon to ischaemia and it might be worth looking at the problems of burning glucose in neurons next. And the problems from failing to generate adequate superoxide for maximal health. There's a lot to think about.
Peter
The first thing to say is that they (ketones) don't seem to work terribly well. I picked up this paper via the Deanna Protocol website. I wrote an unpublished post at the time setting down what an abysmally written paper it is but I thought I would stick to the basics today. Feeding a ketogenic diet to the mice engineered to have an ALS-like disease delays their time to falling off a log but does not extend their lifespan:
"There was no statistically significant difference in the age at death between KD fed animals compared to SOD1-G93 transgenic mice fed a standard laboratory diet (133 ± 4 vs. 131 ± 4 days, p = 0.914)"
Some improved motor function, for a while, may be worth having if you suffer from ALS but I don't think it's exactly a cure or remission.
Although the methods section is very reticent about the diet, it is high in carbohydrate (20% of calories) and protein (20% of calories), so must be MCT based to achieve ketosis.
The findings are confirmed by a nicely written, very clear paper using caprylic acid as a supplement to standard CIAB mouse chow:
"SOD1-G93A animals on caprylic triglyceride diet had a median survival of 135 days. Although it was longer than the median survival of SOD1-G93A animals on control diet (129 days), it did not reach statistical significance (Mantel-Cox test, p=0.165)"
Things weren't much better here in the Deanna Protocol paper. Their ketogenic diet supplied 77% of calories from fat, type unspecified, and essentially zero from carbohydrate. Protein was high at 22% of calories. This is what you get as a result:
"Although the mean survival of SOD1-G93A animals was longer in all three treatment groups than the control group, this difference reached statistical significance only in the KD+DP (4.2%, p = 0.006) and SD+DP groups (7.5%, p = 0.001, Fig. 5, Table 5, Data S2)."
i.e. the ketogenic diet, without the alpha ketoglutarate of the Deanna Protocol, was no better than control mice on CIAB.
All of which is quite interesting and should be quite depressing for groups working with MCTs, ketone esters or ketone salts as managements for neurodegenerative diseases.
We can say certain things about the first two studies. Using MCTs on a moderate to high carbohydrate diet is unlikely to lead to the metabolic changes of a true ketogenic diet. Normoglycaemia is probably not on the menu. It will not lead to the sort of effects of minimal carbohydrate, just adequate protein, very high fat diet. The effect of such a diet has been described as unique.
Of course, a few grams of MCTs on a diet of standard lab chow will generate ketones. That is hardly equivalent to a true ketogenic diet with its reduced glycaemia and basement value insulin levels.
As the paper on the Deanna protocol reports:
"Blood glucose was not significantly different between the diet groups", not exactly what was reported for mice eating D12336.
Ultimately, no one yet appears to have looked at a true ketogenic diet in ALS.
The focus is on the ketones. Ketones are good, but they are not magic. There are people who believe that the ketones themselves are simply a surrogate for very low insulin levels, which is magic (You know who you are Wooo!) and that the benefits of ketogenic diets may stem from the low insulin levels rather than the ketones per se, certainly for obesity management. For neurodegenration I find this idea very appealing. I think that the low glucose/insulin might be particularly important within the brain. I can't see that the work has been done yet, too much of a focus on ketones.
As something of an aside, the Deanna Protocol is interesting in its own right. The core supplement is (arginine-linked) alpha ketoglutarate. From the Protons point of view, if the alpha ketoglutarate enters the TCA at alpha ketoglutarate dehydrogenase and leaves it at malate, it would appear to be a very FADH2 selective input at complex II, generating an NADH:FADH2 ratio of 1:1, i.e. it is functioning as a rather specific FADH2 input. We're all aware that complex I dysfunction is a hallmark of neurodegenerative diseases and, in the absence of beta oxidation (we're in neurons here), complex II is the primary route in to the CoQ couple for electrons via FADH2. Along with mtG3Pdh of course, if that happens to be active. I can see the logic to using this AKG to push complex II without the excess rise in non-usable NADH, which large amounts of acetyl-CoA provide. I'm not surprised AKG is the core component of the Deanna Protocol and hats off to her father for picking this up.
A further aside, Deanna tried coconut oil, caprylic acid and MCTs early on in her disease. Not a lot of help. Adding extra acetyl-CoA from ketones will be of limited help in a condition with complex I dysfunction. My interest still lies in ketones combined with low blood glucose, not as an add on to healthy starches.
It is quite clear from the last post featuring cardiac ischaemia and ketones that any old ketones will do when hypoxia is the problem: Bring on the MCTs. Logically ketones are fats, part pre-oxidised in the liver, so require less oxygen to complete their metabolism in the cardiac muscle. They do not uncouple protons from oxidative phosphorylation either, which we will probably come back to. And while normal fatty acids do uncouple ox phos, this effect is (under normal circumstances) completely lost when mitochondrial ATP levels fall. This probably happens rather quickly under hypoxia.
The energetic failure of neurodegenerative diseases is only partially amenable to ketones. We are looking at a rather different phenomenon to ischaemia and it might be worth looking at the problems of burning glucose in neurons next. And the problems from failing to generate adequate superoxide for maximal health. There's a lot to think about.
Peter
Sunday, September 21, 2014
Should idiots be allowed to write the methods section of any "scientific" paper?
Time to get back to the blog. We had a great summer and life has just kept on being more interesting than blogging. There are about 20 comments I need to read and approve which I'll do my best to get around to, but I thought it was time to hit the keyboard after the summer holidays.
I thought I would just post briefly on the struggles of trying to work out exactly what a given paper is describing in dietary research. I did set out a post a few weeks ago, being rather derogatory, about this paper on ALS. Here is the preamble:
I feel I should like the paper. Really. What with all this iced water being poured over people's heads in the name of ALS research etc. But it's hard.
OK. We're looking at a ketogenic diet for mice endowed with an engineered model of ALS which is quite similar to one of the familial forms of human ALS.
Being me, I go to the methods section first, to see what they fed the poor mice on. From the philosophical point of view I expect the methods section of a paper to allow me to duplicate a given research protocol. All I am told in this case is that the ketogenic diet is 60% fat, 20% carbs and 20% protein and that it was made by Research Diet, Inc. New Brunswick, NJ. That's it. Now, until RD are bought out by some other multinational company, they have a website and this tells me that they supply only one ketogenic diet, D12336, which is 11% protein 89% fat and zero carbohydrate, pretty much what you need to get a rodent in to mild ketosis. So this research group are using a custom diet, what goes in to it is anyone's guess.
My guess is medium chain triglycerides. I don't think you can get a mouse in to ketosis with protein at 20% of calories and carbohydrate at 20%. You'd have trouble getting a human in to ketosis with this, unless you used MCTs.
This is important because I'm interested in teasing out whether there is any point in the enormous effort and endless tedium of eating a low carbohydrate driven ketogenic diet with thyroid deficiency, lethargy, brain fog, glucose deficiency and auto immune disease predisposition as routine sequelae, not to mention the constipation and halitosis (is this me?), when merely popping down to my local Caribbean corner store for a bottle of coconut oil might do the job equally well.
What goes in to the diet matters. Coconut oil is not safflower oil, is not butter. What goes in to the methods matters. Research must be replicable.
End of preamble. I wasn't best impressed.
Before I go on to think about ALS and what help medium chain triglycerides may or may not provide in another post, I thought I would just like to revisit the lethal effects of a VLC keogenic diet paper on the outcome of induced ischaemia and reperfusion of the myocardium in some hapless rats.
It is fairly clear that using "vegetable shortening" as your primary ketogenic source of calories is likely to destroy your myocardium if you have an ischaemic episode. Your first heart attack might be your last. It took an email to Research Diets to get the information about the probable trans fat content of their diet and confirmed to me that the research group had written a methods section which put their paper, and probably the researchers, in to the garbage category.
The same appears to apply to the flip side. I had wondered what a non vegetable shortening based ketogenic diet might do under the same circumstances. Well I'd missed the study, which fully reinforces my pro-LC confirmation bias. Ketogenic diets are the bees-knees for surviving a period of cardiac ischaemia, Crisco excepted.
So, what does the miracle diet for surviving your next coronary look like? I don't know. You don't know. You can read the full text. You still won't know.
The diet is 60% protein by calories! And 10% carbohydrate. The remaining 30% is "oil". Now you know as much as in the paper. Can you replicate the study, based on the methods? No.
BTW: They didn't even check ketone levels! I think we have to assume MCTs again and assume some degree of ketosis.
Crap.
The scrutineers also need to be up against the wall come the revolution.
For all three papers. Crap. Even though I like the results.
Hiya all!
Peter
I thought I would just post briefly on the struggles of trying to work out exactly what a given paper is describing in dietary research. I did set out a post a few weeks ago, being rather derogatory, about this paper on ALS. Here is the preamble:
I feel I should like the paper. Really. What with all this iced water being poured over people's heads in the name of ALS research etc. But it's hard.
OK. We're looking at a ketogenic diet for mice endowed with an engineered model of ALS which is quite similar to one of the familial forms of human ALS.
Being me, I go to the methods section first, to see what they fed the poor mice on. From the philosophical point of view I expect the methods section of a paper to allow me to duplicate a given research protocol. All I am told in this case is that the ketogenic diet is 60% fat, 20% carbs and 20% protein and that it was made by Research Diet, Inc. New Brunswick, NJ. That's it. Now, until RD are bought out by some other multinational company, they have a website and this tells me that they supply only one ketogenic diet, D12336, which is 11% protein 89% fat and zero carbohydrate, pretty much what you need to get a rodent in to mild ketosis. So this research group are using a custom diet, what goes in to it is anyone's guess.
My guess is medium chain triglycerides. I don't think you can get a mouse in to ketosis with protein at 20% of calories and carbohydrate at 20%. You'd have trouble getting a human in to ketosis with this, unless you used MCTs.
This is important because I'm interested in teasing out whether there is any point in the enormous effort and endless tedium of eating a low carbohydrate driven ketogenic diet with thyroid deficiency, lethargy, brain fog, glucose deficiency and auto immune disease predisposition as routine sequelae, not to mention the constipation and halitosis (is this me?), when merely popping down to my local Caribbean corner store for a bottle of coconut oil might do the job equally well.
What goes in to the diet matters. Coconut oil is not safflower oil, is not butter. What goes in to the methods matters. Research must be replicable.
End of preamble. I wasn't best impressed.
Before I go on to think about ALS and what help medium chain triglycerides may or may not provide in another post, I thought I would just like to revisit the lethal effects of a VLC keogenic diet paper on the outcome of induced ischaemia and reperfusion of the myocardium in some hapless rats.
It is fairly clear that using "vegetable shortening" as your primary ketogenic source of calories is likely to destroy your myocardium if you have an ischaemic episode. Your first heart attack might be your last. It took an email to Research Diets to get the information about the probable trans fat content of their diet and confirmed to me that the research group had written a methods section which put their paper, and probably the researchers, in to the garbage category.
The same appears to apply to the flip side. I had wondered what a non vegetable shortening based ketogenic diet might do under the same circumstances. Well I'd missed the study, which fully reinforces my pro-LC confirmation bias. Ketogenic diets are the bees-knees for surviving a period of cardiac ischaemia, Crisco excepted.
So, what does the miracle diet for surviving your next coronary look like? I don't know. You don't know. You can read the full text. You still won't know.
The diet is 60% protein by calories! And 10% carbohydrate. The remaining 30% is "oil". Now you know as much as in the paper. Can you replicate the study, based on the methods? No.
BTW: They didn't even check ketone levels! I think we have to assume MCTs again and assume some degree of ketosis.
Crap.
The scrutineers also need to be up against the wall come the revolution.
For all three papers. Crap. Even though I like the results.
Hiya all!
Peter
Friday, July 11, 2014
Neuron fuel and function
In the comments following a previous post Dustin linked to this rather lovely paper from the early 1970s, back when I was still at school and marathon racing my kayak.
This is one of the nicest figures:

The lower section is the interesting part. CJ had not eaten for 50 days and had a (very) fasting blood glucose of 4.0mmol/l when he received around 0.1 IU/kg of insulin by intravenous injection. Please don't try this at home. You can see that a) he is still insulin sensitive and b) his blood glucose bottomed out at around 0.5mmol/l by 60 minutes. Throughout this period he was asymptomatic. No hypo.
CJ was not running his brain on glucose. The upper section shows a rapid and sustain increase in B-OHB extraction (the dark hatching between the arterial line and jugular bulb line for B-OHB) by his brain through this period of time. Ketones, at levels in excess of 11.0mmol/l, can sustain apparently normal brain function. Given an alternative fuel source this would seem to put the level of blood glucose needed for normal neural function at some (non determined) value of less than 0.5mmol/l.
Drenick et al did not look at fatty acid extraction by the brain. That's a pity, but understandable. No one expects the brain to metabolise palmitic acid. Well, perhaps we should say that neurons should not metabolise palmitic acid. Astrocytes do. Astrocytes are ketogenic and are in a perfect position to supply ketone bodies to neurons using the monocarboxylate transporters ubiquitous on them. I hope to come back to this by the end of the post.
Of course, a 50 day starvation period is not exactly the normal human predicament and we could argue that the normal human brain is glucose dependent. This too, I think may be a very debatable point.
Work has been done with humans under hypoglycaemia with brain function supported by either lactate or pyruvate by intravenous infusion. They're fairly effective, not perfect, but there are limits in how far you can push the metabolism of human volunteers.
Rats are not so fortunate.
If we go to Figure 1 from the rather nice paper emailed to me by Edward

we can see that, under the influence of a massive 20 IU/kg of insulin, there is an almost complete loss of plasma glucose and a slightly more complete loss of brain response to limb stimulus in the insulin-only, profoundly hypoglycaemic group, top row of section A. This has occurred by two and a half hours. The next row has had the hypoglycaemia corrected with glucose (i.e. it's essentially a control group) and has a normal response to stimulus at 4 hours. The lower row shows the effect of lactate in supporting brain function during four hours of persistent, profound, uncorrected hypoglycaemia. You have to note that progress from left to right is time in milliseconds after stimulus and that there is a clear cut delay of about 10 ms in the response time under pure lactate compared to under glucose. This is reiterated in section D, where the response can be seen to be delayed and blunted when compared to the glucose supported data of curve C.
This has led the authors to speculate that, heresy of heresies, there may actually be an absolute need for some glucose by the brain! Strange I know, but... They're not sure of this, just speculating. There are other potential explanations.
Now, most people do not walk around with a blood lactate of 9.0mmol/l. Perhaps most people really do run their brain on glucose?
This seems very unlikely. Or, rather, it seems very unlikely that the neurons in the brain run on glucose. Astrocytes certainly do. But one of the main functions of astrocytes appears to be to manufacture lactate from glucose (directly or from stored glycogen) and deliver it to neurons as a one step conversion fuel giving pyruvate, which can enter the TCA as acetyl-CoA without any messy glycolysis. There is an awful lot of information in this paper.
I put up this nice illustration previously, in the Protons thread:

Neurons are spared glucose. Why?
Mitochondrial glycerol-3-phosphate dehydrogenase. Glucose, during glycolysis, is quite able to input to the electron transport chain through an FADH2 based input at mtG3Pdh, which can reduce the CoQ couple and set the ETC up for reverse electron flow through complex I, with the generation of superoxide as this occurs. Modest superoxide is a Good Thing, especially if you want to signal for mitochondrial biogenesis or cell division. Excess superoxide is a potent signal for apoptosis. Apoptosis is verboten for CNS neurons because any information stored in their synaptic connections will be lost along with the cell. Replacing the cells will hardly replace the memories and appears equally forbidden. There is a need for immortality without reproduction in neurons, for their survival in excess of the lifespan of an organism needing a functional memory in a learning brain.
Feeding lactate through pyruvate to acetyl-CoA does not drive CoQ pool reduction "ahead" of the throughput of electrons coming in from complex I. Neurons do not want to generate insulin resistance. Avoiding glycolysis looks (to me) to be the way they do this. Generating hyperglycaemia looks like a way to overcome the normal lactate shuttle and of forcing glucose directly in to neurons. Enough apoptosis and eventually neural loss just might show as memory loss.
Hyperglycaemia and Alzheimer's...
Generating large amounts of superoxide in astrocytes during glycolysis is not damaging to the neurons supported by the derived lactate. Astrocytes are certainly replaceable, although there seems to be some debate about cell division vs stem cell recruitment. Astrocytes are also able to divide unreasonably rapidly and form various grades of brain tumour. They are common and frequently aggressive. Neuron derived tumours are much rarer and are usually derived from embryonic cells giving medullablastomas rather than being derived from mature neurons. That seems to fit the metabolic arrangements in the brain rather neatly.
This takes us back, eventually, to palmitate as a ketogenic energy supply to the brain via astrocytes. Again, an FADH2 input through electron transporting flavoprotein dehydrogenase can couple with hyperglycaemia to generate reverse electron flow through complex I giving excess superoxide generation. I consider this to be why free fatty acids are excluded from neurons. It's not that FFAs generate excess superoxide per se, they don't. But combined with hyperglycaemia they certainly do, especially palmitate and the longer of the saturated fatty acid series. You really don't want this happening in a cell whose remit is immortality.
Ketones and lactate do not drive reverse electron flow through complex I. Glucose can. Palmitate certainly can. What you want from a metabolic fuel depends on the remit of your cell types. Neurons within the brain preserve information by their continued existence. This is best done by burning lactate or ketones. NOT glucose and, of course, not FFAs. Anyone who claims that glucose is the preferred metabolic fuel of the brain has not though about what a neuron has to do and what an astrocyte actually does do. Or much about the electron transport chain.
Peter
This is one of the nicest figures:

The lower section is the interesting part. CJ had not eaten for 50 days and had a (very) fasting blood glucose of 4.0mmol/l when he received around 0.1 IU/kg of insulin by intravenous injection. Please don't try this at home. You can see that a) he is still insulin sensitive and b) his blood glucose bottomed out at around 0.5mmol/l by 60 minutes. Throughout this period he was asymptomatic. No hypo.
CJ was not running his brain on glucose. The upper section shows a rapid and sustain increase in B-OHB extraction (the dark hatching between the arterial line and jugular bulb line for B-OHB) by his brain through this period of time. Ketones, at levels in excess of 11.0mmol/l, can sustain apparently normal brain function. Given an alternative fuel source this would seem to put the level of blood glucose needed for normal neural function at some (non determined) value of less than 0.5mmol/l.
Drenick et al did not look at fatty acid extraction by the brain. That's a pity, but understandable. No one expects the brain to metabolise palmitic acid. Well, perhaps we should say that neurons should not metabolise palmitic acid. Astrocytes do. Astrocytes are ketogenic and are in a perfect position to supply ketone bodies to neurons using the monocarboxylate transporters ubiquitous on them. I hope to come back to this by the end of the post.
Of course, a 50 day starvation period is not exactly the normal human predicament and we could argue that the normal human brain is glucose dependent. This too, I think may be a very debatable point.
Work has been done with humans under hypoglycaemia with brain function supported by either lactate or pyruvate by intravenous infusion. They're fairly effective, not perfect, but there are limits in how far you can push the metabolism of human volunteers.
Rats are not so fortunate.
If we go to Figure 1 from the rather nice paper emailed to me by Edward
we can see that, under the influence of a massive 20 IU/kg of insulin, there is an almost complete loss of plasma glucose and a slightly more complete loss of brain response to limb stimulus in the insulin-only, profoundly hypoglycaemic group, top row of section A. This has occurred by two and a half hours. The next row has had the hypoglycaemia corrected with glucose (i.e. it's essentially a control group) and has a normal response to stimulus at 4 hours. The lower row shows the effect of lactate in supporting brain function during four hours of persistent, profound, uncorrected hypoglycaemia. You have to note that progress from left to right is time in milliseconds after stimulus and that there is a clear cut delay of about 10 ms in the response time under pure lactate compared to under glucose. This is reiterated in section D, where the response can be seen to be delayed and blunted when compared to the glucose supported data of curve C.
This has led the authors to speculate that, heresy of heresies, there may actually be an absolute need for some glucose by the brain! Strange I know, but... They're not sure of this, just speculating. There are other potential explanations.
Now, most people do not walk around with a blood lactate of 9.0mmol/l. Perhaps most people really do run their brain on glucose?
This seems very unlikely. Or, rather, it seems very unlikely that the neurons in the brain run on glucose. Astrocytes certainly do. But one of the main functions of astrocytes appears to be to manufacture lactate from glucose (directly or from stored glycogen) and deliver it to neurons as a one step conversion fuel giving pyruvate, which can enter the TCA as acetyl-CoA without any messy glycolysis. There is an awful lot of information in this paper.
I put up this nice illustration previously, in the Protons thread:

Neurons are spared glucose. Why?
Mitochondrial glycerol-3-phosphate dehydrogenase. Glucose, during glycolysis, is quite able to input to the electron transport chain through an FADH2 based input at mtG3Pdh, which can reduce the CoQ couple and set the ETC up for reverse electron flow through complex I, with the generation of superoxide as this occurs. Modest superoxide is a Good Thing, especially if you want to signal for mitochondrial biogenesis or cell division. Excess superoxide is a potent signal for apoptosis. Apoptosis is verboten for CNS neurons because any information stored in their synaptic connections will be lost along with the cell. Replacing the cells will hardly replace the memories and appears equally forbidden. There is a need for immortality without reproduction in neurons, for their survival in excess of the lifespan of an organism needing a functional memory in a learning brain.
Feeding lactate through pyruvate to acetyl-CoA does not drive CoQ pool reduction "ahead" of the throughput of electrons coming in from complex I. Neurons do not want to generate insulin resistance. Avoiding glycolysis looks (to me) to be the way they do this. Generating hyperglycaemia looks like a way to overcome the normal lactate shuttle and of forcing glucose directly in to neurons. Enough apoptosis and eventually neural loss just might show as memory loss.
Hyperglycaemia and Alzheimer's...
Generating large amounts of superoxide in astrocytes during glycolysis is not damaging to the neurons supported by the derived lactate. Astrocytes are certainly replaceable, although there seems to be some debate about cell division vs stem cell recruitment. Astrocytes are also able to divide unreasonably rapidly and form various grades of brain tumour. They are common and frequently aggressive. Neuron derived tumours are much rarer and are usually derived from embryonic cells giving medullablastomas rather than being derived from mature neurons. That seems to fit the metabolic arrangements in the brain rather neatly.
This takes us back, eventually, to palmitate as a ketogenic energy supply to the brain via astrocytes. Again, an FADH2 input through electron transporting flavoprotein dehydrogenase can couple with hyperglycaemia to generate reverse electron flow through complex I giving excess superoxide generation. I consider this to be why free fatty acids are excluded from neurons. It's not that FFAs generate excess superoxide per se, they don't. But combined with hyperglycaemia they certainly do, especially palmitate and the longer of the saturated fatty acid series. You really don't want this happening in a cell whose remit is immortality.
Ketones and lactate do not drive reverse electron flow through complex I. Glucose can. Palmitate certainly can. What you want from a metabolic fuel depends on the remit of your cell types. Neurons within the brain preserve information by their continued existence. This is best done by burning lactate or ketones. NOT glucose and, of course, not FFAs. Anyone who claims that glucose is the preferred metabolic fuel of the brain has not though about what a neuron has to do and what an astrocyte actually does do. Or much about the electron transport chain.
Peter
Wednesday, June 25, 2014
Cardiac ischaemia and low carbohydrate diets
There are researchers within the worlds of both nutrition and cardiology who appear to be very determined to supply a message that failing to eat adequate carbohydrate is a very dangerous choice. They occasionally produce little gems of research.
This paper, which has been sitting on my hard drive for a few years, surfaced on Facebook recently. I gather it has been cited by someone who’s writing requires more ondansetron to read than I currently possess. I’ll just assume it was some sort of “eat LC and the first smidgin of myocardial hypoxia will finish you off” warning, but I’m just guessing and I have every intention of keeping it that way.
The paper itself is very convincing, well written and the protocol extensively justified. The core findings are that a LC diet impairs insulin signalling, depletes myocardial glycogen and results in massive necrosis during reperfusion after a period of myocardial hypoxia. The basic idea is that the lack of glycogen limits substrate for anaerobic glycolysis and failed insulin signalling both impairs glucose delivery from the perfusate and also fails to deliver a number of highly beneficial insulin effects which are independent of GLUT4 translocation.
This is the fate of the LC myocardium. As one of my co workers might say: Dig the hole, choose the coffin.

Obviously, for a LC eater, this is disturbing. The queue for McDougall-ism is over there.
The first thing which I find slightly disturbing is that, in a trial of the Atkins Diet™ (always mention by name), the rats ate more and were significantly heavier than the control rats, within two weeks. I’m not totally certain if I remember correctly, but I thought that the Atkins Diet was used for weight LOSS, not weight gain. Perhaps the authors might have been a little disturbed by this finding too, but apparently it doesn’t need mention. Hmmm.
The Atkins™ like diet is TestDiet 5TSY, no longer manufactured. Table I is abstracted by the authors from the full formula provided by Purina and supplies information on a need-to-know basis.

So we know total fat, saturated fat and that “The diets have the same concentrations of essential fatty acids”, i.e. we don’t quite know what the diets were made of. But Purina still will email you a pdf (very promptly) and that gives you this:

There is no mention of Crisco™ by name and no information about the trans fat content, but can you guess how much "Vegetable shortening" is in the control diet? Oh, you guessed!
I know it seems stupid to say this, but if you want to lose weight and/or survive a heart attack, for goodness sake do not choose Crisco or its equivalent as 20% of your calorie intake.
Now, this study does not tell us that a low carbohydrate diet is is good or bad for surviving a heart attack, it's simply not possible to pull that information out due to the lack of control of the variables in the diets. One can only wonder whether the formula of the Atkins Rodent Diet was specifically developed to cause metabolic problems or that somewhere along the line the original Atkins diet suggested a generous consumption of trans fat based vegetable shortening. Maybe I missed this.
I guess you could leave it there and say don't eat vegetable shortening, but there are a whole stack of follow on ideas to this study. There is MASSIVE cardiac necrosis in the LC group of rats. If it is the trans fats, how do they cause this?
One very interesting aspect of the study is the pre-ischaemia depletion of glycogen. These rats are not in ketosis. The doubling of ketone levels from 0.3mmol/l to 0.6mmol/l may well be statistically significant, but is not biologically significant. To go back, yet again, to Veech et al, we need around 5.0mmol/l of mixed ketone bodies to completely replace the insulin signalling system. You can't sidestep insulin resistance with 0.6mmol/l B-OHB. You simply cannot get rats in to a functional level of ketosis with protein at 30% of calories and carbohydrate at 12%. You need carbs near zero and protein limited to < 10% of calories to have rats in nutritional ketosis. Some papers limit protein to < 5% of calories, with minimal carbohydrate.
A period of starvation for rats on the control chow would have tested the hypothesis that it was a lack of glycogen which damaged the myocardium under hypoxia.
My own idea is that the glycogen depletion might be a surrogate for insulin resistance rather than carbohydrate restriction. We have long known that trans fatty acids from partially hydrogenated vegetable oils induce insulin resistance (though not in every study!).
So the next thought is: What sort of insulin resistance?
Are we thinking about an excess of superoxide from complex I, to imitate post prandial hyper caloric insulin resistance? Or are we thinking about the insulin resistance of fasting, when uncoupling results in blunted insulin signalling combined with high oxygen consumption and limited ATP generation?
I like the second idea when applied to this study.
If trans fatty acids, which are structurally similar but not quite identical to saturated fatty acids, allow persistent low level uncoupling outside of the physiological role of the normally structured FFAs it is interesting to speculate that they may continue to allow uncoupling when uncoupling should be utterly and totally banned.
The consequences would be decreased ATP production per unit oxygen consumed. A bit like the findings in Table 3 of this paper. The last column gives you the ATP generated per unit O2 consumed. This would be a disaster under hypoxic conditions. The IF line uses industrial trans fats. The soleus muscle is the mitochondrial dependent one cf the tibialis muscle..

ASIDE: It's worth noting that cytoplasmic ATP is a marked inhibitor of uncoupling but that this is easily overcome by adequate levels of mitochondrial ATP binding within the UCP pore from the end which does not produce a conformational change. Figure 7 gives the details but the main text is really clever stuff (without an agenda, as far as I can see). I'll blog about this paper sometime.

Under this control system the occurrence of falling mitochondrial ATP levels should allow cytoplasmic ATP to immediately shut down the normal uncoupling associated with ketogenic eating and maximise coupled ATP production per unit oxygen when this is needed. The interesting question is whether the uncoupling suspected of transfats persists under low mitochondrial ATP levels. END ASIDE.
You could speculate for hours about what trans fats may or may not do.
This is a very fertile area for idea generation. Ultimately, we don't know what a LC diet based on real Food would do to ischaemic damage in the heart. Maybe it will be as bad as a trans fat based LC (weight gain inducing) diet, maybe less severe.
We are never going to find out what Food does by using "AIN-93G Atkins/Rodent 5TSY" diet based experiments. The rats died in vain, especially as the study buried the possibility of lethal effects from trans fats.
Peter
This paper, which has been sitting on my hard drive for a few years, surfaced on Facebook recently. I gather it has been cited by someone who’s writing requires more ondansetron to read than I currently possess. I’ll just assume it was some sort of “eat LC and the first smidgin of myocardial hypoxia will finish you off” warning, but I’m just guessing and I have every intention of keeping it that way.
The paper itself is very convincing, well written and the protocol extensively justified. The core findings are that a LC diet impairs insulin signalling, depletes myocardial glycogen and results in massive necrosis during reperfusion after a period of myocardial hypoxia. The basic idea is that the lack of glycogen limits substrate for anaerobic glycolysis and failed insulin signalling both impairs glucose delivery from the perfusate and also fails to deliver a number of highly beneficial insulin effects which are independent of GLUT4 translocation.
This is the fate of the LC myocardium. As one of my co workers might say: Dig the hole, choose the coffin.

Obviously, for a LC eater, this is disturbing. The queue for McDougall-ism is over there.
The first thing which I find slightly disturbing is that, in a trial of the Atkins Diet™ (always mention by name), the rats ate more and were significantly heavier than the control rats, within two weeks. I’m not totally certain if I remember correctly, but I thought that the Atkins Diet was used for weight LOSS, not weight gain. Perhaps the authors might have been a little disturbed by this finding too, but apparently it doesn’t need mention. Hmmm.
The Atkins™ like diet is TestDiet 5TSY, no longer manufactured. Table I is abstracted by the authors from the full formula provided by Purina and supplies information on a need-to-know basis.
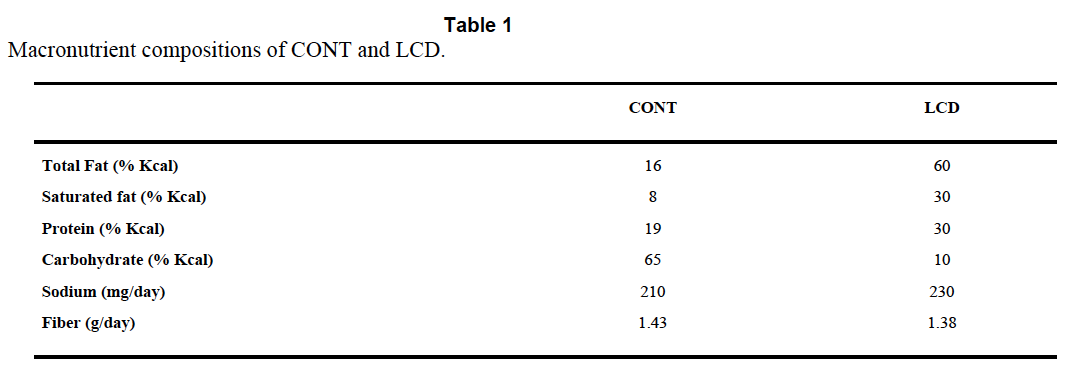
So we know total fat, saturated fat and that “The diets have the same concentrations of essential fatty acids”, i.e. we don’t quite know what the diets were made of. But Purina still will email you a pdf (very promptly) and that gives you this:

There is no mention of Crisco™ by name and no information about the trans fat content, but can you guess how much "Vegetable shortening" is in the control diet? Oh, you guessed!
I know it seems stupid to say this, but if you want to lose weight and/or survive a heart attack, for goodness sake do not choose Crisco or its equivalent as 20% of your calorie intake.
Now, this study does not tell us that a low carbohydrate diet is is good or bad for surviving a heart attack, it's simply not possible to pull that information out due to the lack of control of the variables in the diets. One can only wonder whether the formula of the Atkins Rodent Diet was specifically developed to cause metabolic problems or that somewhere along the line the original Atkins diet suggested a generous consumption of trans fat based vegetable shortening. Maybe I missed this.
I guess you could leave it there and say don't eat vegetable shortening, but there are a whole stack of follow on ideas to this study. There is MASSIVE cardiac necrosis in the LC group of rats. If it is the trans fats, how do they cause this?
One very interesting aspect of the study is the pre-ischaemia depletion of glycogen. These rats are not in ketosis. The doubling of ketone levels from 0.3mmol/l to 0.6mmol/l may well be statistically significant, but is not biologically significant. To go back, yet again, to Veech et al, we need around 5.0mmol/l of mixed ketone bodies to completely replace the insulin signalling system. You can't sidestep insulin resistance with 0.6mmol/l B-OHB. You simply cannot get rats in to a functional level of ketosis with protein at 30% of calories and carbohydrate at 12%. You need carbs near zero and protein limited to < 10% of calories to have rats in nutritional ketosis. Some papers limit protein to < 5% of calories, with minimal carbohydrate.
A period of starvation for rats on the control chow would have tested the hypothesis that it was a lack of glycogen which damaged the myocardium under hypoxia.
My own idea is that the glycogen depletion might be a surrogate for insulin resistance rather than carbohydrate restriction. We have long known that trans fatty acids from partially hydrogenated vegetable oils induce insulin resistance (though not in every study!).
So the next thought is: What sort of insulin resistance?
Are we thinking about an excess of superoxide from complex I, to imitate post prandial hyper caloric insulin resistance? Or are we thinking about the insulin resistance of fasting, when uncoupling results in blunted insulin signalling combined with high oxygen consumption and limited ATP generation?
I like the second idea when applied to this study.
If trans fatty acids, which are structurally similar but not quite identical to saturated fatty acids, allow persistent low level uncoupling outside of the physiological role of the normally structured FFAs it is interesting to speculate that they may continue to allow uncoupling when uncoupling should be utterly and totally banned.
The consequences would be decreased ATP production per unit oxygen consumed. A bit like the findings in Table 3 of this paper. The last column gives you the ATP generated per unit O2 consumed. This would be a disaster under hypoxic conditions. The IF line uses industrial trans fats. The soleus muscle is the mitochondrial dependent one cf the tibialis muscle..
ASIDE: It's worth noting that cytoplasmic ATP is a marked inhibitor of uncoupling but that this is easily overcome by adequate levels of mitochondrial ATP binding within the UCP pore from the end which does not produce a conformational change. Figure 7 gives the details but the main text is really clever stuff (without an agenda, as far as I can see). I'll blog about this paper sometime.

Under this control system the occurrence of falling mitochondrial ATP levels should allow cytoplasmic ATP to immediately shut down the normal uncoupling associated with ketogenic eating and maximise coupled ATP production per unit oxygen when this is needed. The interesting question is whether the uncoupling suspected of transfats persists under low mitochondrial ATP levels. END ASIDE.
You could speculate for hours about what trans fats may or may not do.
This is a very fertile area for idea generation. Ultimately, we don't know what a LC diet based on real Food would do to ischaemic damage in the heart. Maybe it will be as bad as a trans fat based LC (weight gain inducing) diet, maybe less severe.
We are never going to find out what Food does by using "AIN-93G Atkins/Rodent 5TSY" diet based experiments. The rats died in vain, especially as the study buried the possibility of lethal effects from trans fats.
Peter
Monday, June 16, 2014
Slim mice which don't fart
Germ free mice are quite interesting. I suppose that the first thing we can say about them is that they don’t have any bacterial fermentation in their gut to produce flatus. As a side issue of some interest is that none of their intestinal mucosal cells ever sees any acetate, butyrate or propionate derived from microbial fermentation of fibre in the gut. They seem rather happy that way. You could also say the same about their liver, it too never sees any SCFA bacterial fermentation products. They stay slim.
Ancient history tells us that germ free mice live rather longer than their more flatulent counterparts. I find this quite interesting.
Of course longevity is a relative term and perhaps ought to be qualified a little. We should actually say that they live longer than conventional mice, provided you feed them. Not feeding germ free mice is quite bad for them, they die under starvation significantly sooner than conventional mice do, at higher bodyweight and with more fat reserves.
That was the state of our knowledge at the end of the last century.
In more recent years the mechanisms for this failure to cope with starvation has become a little clearer. Germ free mice are insulin sensitive. They stay that way pretty much whatever you do to them in terms of diet. They stay that way even if you starve them. That, in terms of survival, is a booboo.
There are at least two techniques available via the gut microbiota which might improve the ability to survive starvation and which are gifted to germ free mice by smearing them with pooh from a conventional mouse. One is endotoxin, a subject I suspect I will come back to. The current one, for this post, is short chain fatty acids.
Bacterial fermentation of fibre produces those miracle agents of gut health and general goodness; acetate, butyrate and propionate. These act through a G protein coupled receptor on enterendocrine cells to promote fat storage. This is the Gpr41 receptor. Needless to say the enteroendocrine cells are the same cells which secrete FIAF, as in the FIAF series of posts. The two are possibly related. Given a little effort we could, by looking at conventionalisation of germ free mice, make a good guess about how much of a mouse’s fat belongs to the mouse and how much belongs to its gut microbiota.
So the fermentation products of bacteria promote fat storage when germ free mice are conventionalised. But no one seems to think that a lack of fat was the reason for germ free mice dying sooner under starvation. So what is the other effect of SCFAs, other than a bigger butt?
The gift of ketosis. Germ free mice are crap at ketosis. It’s not that they can’t do it, it seems to be more like a lack of practice. Acetate and butyrate are particularly ketogenic and hit all sorts of signalling systems in the liver to up regulate ketone generation in conventional mice. Of course germ free mice on standard crapinabag never send acetate to their liver, so their liver never up-regulates the correct PPARs to do ketosis. Putting germ free mice on to a deeply, deeply ketogenic diet teaches them how to make ketones and they become rather good at it.
Under starvation the myocardium of a germ free mouse continues to metabolise glucose, despite free fatty acids being available. Crawford et al consider the myocardium to be fairly representative of many of the glucose using organs in the body. They view ketones as an alternative energy source to glucose under starvation. While no one would argue with this, the possibility which fascinates me is that ketones are turning off glycolysis to spare glucose for the brain when fatty acids alone don’t do this. They appear to be a core component of physiological insulin resistance.
Whether these ketogenic germ free mice are able to extend their time of death to that of starved conventional mice is not a question which any modern ethics committee will allow you to answer today, unless you have a damned good reason. But I suspect the answer is yes.
Peter
Addendum. If you accept that perhaps SCFAs expand your butt via Gpr41 (we are not, after all, germ free mice freshly smeared with conventional mouse faeces) guess which metabolite is a direct antagonist to SCFAs at Gpr41? Clue, it's that the beta hydroxylated derivative of butyric acid. I love stuff that makes sense.
Ancient history tells us that germ free mice live rather longer than their more flatulent counterparts. I find this quite interesting.
Of course longevity is a relative term and perhaps ought to be qualified a little. We should actually say that they live longer than conventional mice, provided you feed them. Not feeding germ free mice is quite bad for them, they die under starvation significantly sooner than conventional mice do, at higher bodyweight and with more fat reserves.
That was the state of our knowledge at the end of the last century.
In more recent years the mechanisms for this failure to cope with starvation has become a little clearer. Germ free mice are insulin sensitive. They stay that way pretty much whatever you do to them in terms of diet. They stay that way even if you starve them. That, in terms of survival, is a booboo.
There are at least two techniques available via the gut microbiota which might improve the ability to survive starvation and which are gifted to germ free mice by smearing them with pooh from a conventional mouse. One is endotoxin, a subject I suspect I will come back to. The current one, for this post, is short chain fatty acids.
Bacterial fermentation of fibre produces those miracle agents of gut health and general goodness; acetate, butyrate and propionate. These act through a G protein coupled receptor on enterendocrine cells to promote fat storage. This is the Gpr41 receptor. Needless to say the enteroendocrine cells are the same cells which secrete FIAF, as in the FIAF series of posts. The two are possibly related. Given a little effort we could, by looking at conventionalisation of germ free mice, make a good guess about how much of a mouse’s fat belongs to the mouse and how much belongs to its gut microbiota.
So the fermentation products of bacteria promote fat storage when germ free mice are conventionalised. But no one seems to think that a lack of fat was the reason for germ free mice dying sooner under starvation. So what is the other effect of SCFAs, other than a bigger butt?
The gift of ketosis. Germ free mice are crap at ketosis. It’s not that they can’t do it, it seems to be more like a lack of practice. Acetate and butyrate are particularly ketogenic and hit all sorts of signalling systems in the liver to up regulate ketone generation in conventional mice. Of course germ free mice on standard crapinabag never send acetate to their liver, so their liver never up-regulates the correct PPARs to do ketosis. Putting germ free mice on to a deeply, deeply ketogenic diet teaches them how to make ketones and they become rather good at it.
Under starvation the myocardium of a germ free mouse continues to metabolise glucose, despite free fatty acids being available. Crawford et al consider the myocardium to be fairly representative of many of the glucose using organs in the body. They view ketones as an alternative energy source to glucose under starvation. While no one would argue with this, the possibility which fascinates me is that ketones are turning off glycolysis to spare glucose for the brain when fatty acids alone don’t do this. They appear to be a core component of physiological insulin resistance.
Whether these ketogenic germ free mice are able to extend their time of death to that of starved conventional mice is not a question which any modern ethics committee will allow you to answer today, unless you have a damned good reason. But I suspect the answer is yes.
Peter
Addendum. If you accept that perhaps SCFAs expand your butt via Gpr41 (we are not, after all, germ free mice freshly smeared with conventional mouse faeces) guess which metabolite is a direct antagonist to SCFAs at Gpr41? Clue, it's that the beta hydroxylated derivative of butyric acid. I love stuff that makes sense.
Sunday, June 15, 2014
Cholesterol: Do chylomicrons clog your arteries? (2)
I'll keep this brief.
The comments on the last post are awash with people trying to help my resident lipophobe out of his lipophobia. This is admirable but misguided and ultimately doomed.
Many years ago I looked at the Copenhagen Heart Study. This observational study generated two hypotheses. One is that chylomicrons kill you. That seems enough for a lipophobe and, if you have this mindset, for goodness sake add carbs to your diet and avoid post prandial hyperlipidaemia. The choice is yours. Go for it. The lack of stress will help you no end.
My own hypothesis is that elevated post prandial triglycerides, here in a population on a mixed diet, is a surrogate for insulin resistance. That is rather similar to an elevated HbA1c under the same circumstances. These are both superficial markers for a failure of complex I of the electron transport chain to effectively deal with the amount of NADH being provided by processing of the diet.
The solution from a Hyperlipid point of view is to concentrate on supplying FADH2 to the ETC via beta oxidation of maximally saturated fatty acids and to minimise NADH input via chronic normoglycaemia, possibly assisted by the inhibition of glycolysis by ketones.
I have no interest in converting lipophobes to lipophiles. We all have our problems, we just have to live with them.
But some gems are coming out of chylomicrons and endotoxin reading. I especially enjoyed this one (among many), all quotes from the same paper:
"Therefore, on the basis of current information, lipoproteins modulate the host response to endotoxin by inhibiting the activation of macrophages, monocytes, and other LPS-responsive cells; promoting the catabolism of LPS by the hepatic parenchymal cells; and inhibiting the response of hepatocytes to pro inflammatory stimuli"
"Early in the course of this work, we found that chylomicron increases the clearance of LPS by the liver while decreasing overall TNF-α production"
"These most recent studies are focused on cells that express the low-density lipoprotein receptor and are critical to the innate immune response to infection, including adrenocortical cells and vascular endothelial cells. The postulated series of events, whereby a foreign molecule (i.e., LPS) serves to both trigger and attenuate a programmed cellular stress response, is unprecedented"
My emphasis.
Now, a functional LDL-C receptor is utterly necessary for the anti-inflammatory effect of the chylomicron/endotoxin complex. If someone's ears don't prick up on this one they have clearly never heard of homozygous familial hypecholesterolaemia. The focus on elevated lipids (sigh) might just have missed the core of the problem, which is the failure to internalise lipoproteins. Interesting idea? It certainly is to me.
Peter
The comments on the last post are awash with people trying to help my resident lipophobe out of his lipophobia. This is admirable but misguided and ultimately doomed.
Many years ago I looked at the Copenhagen Heart Study. This observational study generated two hypotheses. One is that chylomicrons kill you. That seems enough for a lipophobe and, if you have this mindset, for goodness sake add carbs to your diet and avoid post prandial hyperlipidaemia. The choice is yours. Go for it. The lack of stress will help you no end.
My own hypothesis is that elevated post prandial triglycerides, here in a population on a mixed diet, is a surrogate for insulin resistance. That is rather similar to an elevated HbA1c under the same circumstances. These are both superficial markers for a failure of complex I of the electron transport chain to effectively deal with the amount of NADH being provided by processing of the diet.
The solution from a Hyperlipid point of view is to concentrate on supplying FADH2 to the ETC via beta oxidation of maximally saturated fatty acids and to minimise NADH input via chronic normoglycaemia, possibly assisted by the inhibition of glycolysis by ketones.
I have no interest in converting lipophobes to lipophiles. We all have our problems, we just have to live with them.
But some gems are coming out of chylomicrons and endotoxin reading. I especially enjoyed this one (among many), all quotes from the same paper:
"Therefore, on the basis of current information, lipoproteins modulate the host response to endotoxin by inhibiting the activation of macrophages, monocytes, and other LPS-responsive cells; promoting the catabolism of LPS by the hepatic parenchymal cells; and inhibiting the response of hepatocytes to pro inflammatory stimuli"
"Early in the course of this work, we found that chylomicron increases the clearance of LPS by the liver while decreasing overall TNF-α production"
"These most recent studies are focused on cells that express the low-density lipoprotein receptor and are critical to the innate immune response to infection, including adrenocortical cells and vascular endothelial cells. The postulated series of events, whereby a foreign molecule (i.e., LPS) serves to both trigger and attenuate a programmed cellular stress response, is unprecedented"
My emphasis.
Now, a functional LDL-C receptor is utterly necessary for the anti-inflammatory effect of the chylomicron/endotoxin complex. If someone's ears don't prick up on this one they have clearly never heard of homozygous familial hypecholesterolaemia. The focus on elevated lipids (sigh) might just have missed the core of the problem, which is the failure to internalise lipoproteins. Interesting idea? It certainly is to me.
Peter
Thursday, June 12, 2014
Endotoxin Absorption on a High Fat Diet
Housekeeping: Endotoxin is always of some interest to an anaesthetist and the link between endotoxin uptake and high fat diets has been in my thoughts somewhat in recent months. This probably ties in to resistant starch in some way, so the next few posts will probably be random doodles around this subject. And maybe why the Protons thread is fairly well out of data to extend it and another look at brain metabolism, particularly why it doesn't run on fat (or glucose much of the time), all need posts as the time becomes available. Plus Toxic forwarded me a link on B12/folate guidelines which can be downloaded here. As she says, medics may well not read these sorts of guidelines but informed patients certainly should. OK here is the post as was planned.
Endotoxin is nasty stuff. If we want some sort of idea of how nasty it is we just have to look at this juicy quote from Hansen et al from Denmark:
“Harte et al. have reported levels of endotoxin to be between 3.3 and 14.2 EU/mL, but these concentrations are known to increase levels of tumor necrosis factor α (TNFα) in plasma and induce a massive inflammatory response in humans”.
The context of the quote is that these folks are commenting on a paper by Harte et al which described the above levels of endotoxin as being found in human volunteers, which should be nearly lethal. The victims, poor folks, had been coerced in to drinking the sort of amount of whipping cream which might be a lunchtimee snack for myself. And they survived. As do I after such an insult (so far).
What galls Hansen appears to be complete lack of any sort of an inflammatory response to this lethal chylomicron/endotoxin cocktail coursing through the bloodstream of the lipophiles (assuming they enjoyed being paid to drink the cream).
In every group studied, from normal through obese and IGT to diabetic, TNFa falls after a decent glass of whipping cream, albeit in a statistically non significant manner. Endotoxin levels go up, often dramatically and highly significantly.
It’s all in Table 2:

Even in the deepest cesspits of nutritional research no one really expects a half a pint of cream to produce lethal endotoxaemia within a few hours of drinking it. In their response to the comment, which implies that they can’t measure endotoxin correctly, Harte are polite and point out that there is a world of difference between an iv bolus of neat endotoxin (not recommended) and the absorption of endotoxin from the gut in the presence of chylomicrons (welcome to my world).
There are a number of papers showing that apolipoprotein B containing lipid particles are markedly protective against endotoxin, albeit mostly in mouse models. Quite why an oral lipid bolus should automatically load chylomicrons with endotoxin in healthy individuals is another of those fascinating questions which may need a little thought but I think we can reliably say that it would have been eliminated if there is no benefit.
I wrote a post, several years ago, where Greve’s group demonstrated a marked protective effect of a gastric lipid gavage against the effects of haemorrhagic shock, the effects of which are largely related to loss of gut wall integrity, bacterial translocation and endotoxin uptake.
In the high fat world, endotoxin uptake is not what it seems. Lipoproteins neutralise and carry endotoxin. If you need systemic inflammation to maintain research funding you need to be a little more McDevious.
The above little exchange came to mind because Zachary mentioned (in comments on the last post) a study at his university recruiting victims to demonstrate endotoxin uptake after a high fat meal WITH inflammation. You can, of course do this. Technique: "The diet is foods such as TV dinners, meat and cheese". This may be good for getting the right result to secure subsequent funding but it obliterates the fact that a single high fat meal of unadulterated whipping cream is both anti inflammatory and might even improve insulin sensitivity, in the acute setting at least. Just skip the Egg McMuffins.
Peter
Endotoxin is nasty stuff. If we want some sort of idea of how nasty it is we just have to look at this juicy quote from Hansen et al from Denmark:
“Harte et al. have reported levels of endotoxin to be between 3.3 and 14.2 EU/mL, but these concentrations are known to increase levels of tumor necrosis factor α (TNFα) in plasma and induce a massive inflammatory response in humans”.
The context of the quote is that these folks are commenting on a paper by Harte et al which described the above levels of endotoxin as being found in human volunteers, which should be nearly lethal. The victims, poor folks, had been coerced in to drinking the sort of amount of whipping cream which might be a lunchtimee snack for myself. And they survived. As do I after such an insult (so far).
What galls Hansen appears to be complete lack of any sort of an inflammatory response to this lethal chylomicron/endotoxin cocktail coursing through the bloodstream of the lipophiles (assuming they enjoyed being paid to drink the cream).
In every group studied, from normal through obese and IGT to diabetic, TNFa falls after a decent glass of whipping cream, albeit in a statistically non significant manner. Endotoxin levels go up, often dramatically and highly significantly.
It’s all in Table 2:

Even in the deepest cesspits of nutritional research no one really expects a half a pint of cream to produce lethal endotoxaemia within a few hours of drinking it. In their response to the comment, which implies that they can’t measure endotoxin correctly, Harte are polite and point out that there is a world of difference between an iv bolus of neat endotoxin (not recommended) and the absorption of endotoxin from the gut in the presence of chylomicrons (welcome to my world).
There are a number of papers showing that apolipoprotein B containing lipid particles are markedly protective against endotoxin, albeit mostly in mouse models. Quite why an oral lipid bolus should automatically load chylomicrons with endotoxin in healthy individuals is another of those fascinating questions which may need a little thought but I think we can reliably say that it would have been eliminated if there is no benefit.
I wrote a post, several years ago, where Greve’s group demonstrated a marked protective effect of a gastric lipid gavage against the effects of haemorrhagic shock, the effects of which are largely related to loss of gut wall integrity, bacterial translocation and endotoxin uptake.
In the high fat world, endotoxin uptake is not what it seems. Lipoproteins neutralise and carry endotoxin. If you need systemic inflammation to maintain research funding you need to be a little more McDevious.
The above little exchange came to mind because Zachary mentioned (in comments on the last post) a study at his university recruiting victims to demonstrate endotoxin uptake after a high fat meal WITH inflammation. You can, of course do this. Technique: "The diet is foods such as TV dinners, meat and cheese". This may be good for getting the right result to secure subsequent funding but it obliterates the fact that a single high fat meal of unadulterated whipping cream is both anti inflammatory and might even improve insulin sensitivity, in the acute setting at least. Just skip the Egg McMuffins.
Peter
Saturday, May 31, 2014
HbA1c: Low glucose and acid (palmitic)
If anyone would like to combine a look at the pathology of low blood glucose levels with the psychology of diabetes research they might do worse than to look at this paper.
Hypoglycaemia is bad, very bad. At the vascular endothelium level and at the mitochondrial delta psi level. The sort of low levels of blood glucose frequently visited by people like Steve Cooksey and myself are a complete disaster. The group is looking at levels of blood glucose that I can easily achieve by skipping a couple of meals combined with a little mild aerobic exercise, walking or cutting the lawn. I’ve seen 2.7mmol/l on my (admittedly relatively inaccurate) Freestyle Lite. I felt fine at the time and I did nothing to adjust my blood glucose level before having supper a few hours later...
Perhaps I should be carb loading, just a little, to avoid hypoglycaemia induced damage? However this might drop my free fatty acid levels, which I would prefer to avoid.
The abstract of any paper is word limited and the role of FFAs in the findings don't get a mention, so you might assume FFAs have no significant bearing on hypoglycaemic injury.
The discussion section of the paper tends to confirm this:
“Our findings with respect to fatty acid and L-carnitine suggest mechanistically that free fatty acid utilization in vivo would not be sufficient to suppress LG induced NO suppression and excessive mitochondrial superoxide production…”
They seem pretty certain, but do they have data to support this? Flicking to the results section we see that the group looked at a palmitate with L-carnitine combination and found that it worked beautifully:
“We found that combined treatment of endothelial cells with 1% palmitate and L-carnitine reduced LG-induced mitochondrial superoxide production to normal levels (Supplemental Figure IIIa)”
Want the picture?

I think we will all agree that the combination of palmitate with L-carnitine completely, totally normalises superoxide production in the low glucose cells and was utterly harmless in the normal glucose cells. The graph speaks against the discussion.
This normalisation of superoxide generation appears to have been too much to bear, the concept that palmitic acid might be beneficial, so they separated the palmitate from the L-carnitine and tried them separately.
“mitochondrial superoxide production increased under LG conditions with the addition of 1% palmitate while L-carnitine alone returned mitochondrial superoxide levels to those similar to the NG condition (Supplemental Figure IIIb)”
Now, this sticks in my craw. This is wrong. Completely.
Here is Supplemental figure IIIb.

Any reading with an * above it is higher than the normal glucose (NG) reading. This includes the low glucose plus L-carnitine. The low glucose with carnitine value is greater than the normal glucose value, p < 0.05.
There is, however, a crucifix above the LG+LC. The superoxide production here is significantly less than that in the LG group with added neat palmitate, that’s the meaning of the crucifix. But because of the asterix we KNOW that the superoxide here is still higher than the normal glucose value, p < 0.05.
Stating that
“L-carnitine alone returned mitochondrial superoxide levels to those similar to the NG condition”
would require that you (could) remove the asterisk from the low glucose plus carnitine column.
You can't.
You might just as well say the normal glucose plus palmitate (NG+palm) is "similar to the NG condition". Which they don't. Asterisk.
Sigh. End rant.
There are many other things which are both good and bad about this paper. But, as a lipophile living in what sometimes feels like a sea of lipophobia, it really ticks me off to see frankly incorrect and unsupported statements like this. I still scratch my head about motive.
It’s quite clear that palmitate/L-carnitine normalised the excess superoxide production of low glucose completely. I have lots of palmitate in my blood stream, though perhaps not the 1% chemical grade palmitic acid used here! I've probably got reasonable amounts of L-carnitine in my cells too, being a moderate meat eater and highly fat adapted. But the big giggle would really have been to use ketones in the study. I have ketones. As Veech pointed out, many years ago now, ketones can completely replace the whole glucose/insulin metabolic pathway in an isolated rat myocardium. Very effectively.
The flip side is that the paper is looking for information about insulin overdose rather than simple low-normal glycaemia. My BG of 2.7mmol/l was in the presence of utterly basal insulin, markedly elevated free fatty acids and modestly elevated ketones. A diabetic on a sugar based diet having a little (or even quite big) accident with their insulin to starch balance will undoubtedly have suppressed FFAs waaaaay before they hypoed and will probably have a metabolism that can't remember what a ketone body is. The metabolic milieu of parenteral insulin overdose is radically different to that of ketogenic eating. You have to engage thought processes before deciding that any blood glucose below 5mmol/l produces graded superoxide overproduction and failure of nitric oxide signalling. It's understandable from a modern diabetologist point of view, just wrong to bury data which do not support your preconceptions.
If non-injecting (and non sulphonylurea popping) folks want to reduce their FFAs and eliminate ketones in order to avoid low-normal glucose levels by eating a few extra grams of carbs, that is absolutely fine. We can all ignore the biochemistry if we so wish, serious researchers do it all the time.
Personally, I still like to have a few ketones around. And rather a lot of free fatty acids.
Peter
Hypoglycaemia is bad, very bad. At the vascular endothelium level and at the mitochondrial delta psi level. The sort of low levels of blood glucose frequently visited by people like Steve Cooksey and myself are a complete disaster. The group is looking at levels of blood glucose that I can easily achieve by skipping a couple of meals combined with a little mild aerobic exercise, walking or cutting the lawn. I’ve seen 2.7mmol/l on my (admittedly relatively inaccurate) Freestyle Lite. I felt fine at the time and I did nothing to adjust my blood glucose level before having supper a few hours later...
Perhaps I should be carb loading, just a little, to avoid hypoglycaemia induced damage? However this might drop my free fatty acid levels, which I would prefer to avoid.
The abstract of any paper is word limited and the role of FFAs in the findings don't get a mention, so you might assume FFAs have no significant bearing on hypoglycaemic injury.
The discussion section of the paper tends to confirm this:
“Our findings with respect to fatty acid and L-carnitine suggest mechanistically that free fatty acid utilization in vivo would not be sufficient to suppress LG induced NO suppression and excessive mitochondrial superoxide production…”
They seem pretty certain, but do they have data to support this? Flicking to the results section we see that the group looked at a palmitate with L-carnitine combination and found that it worked beautifully:
“We found that combined treatment of endothelial cells with 1% palmitate and L-carnitine reduced LG-induced mitochondrial superoxide production to normal levels (Supplemental Figure IIIa)”
Want the picture?

I think we will all agree that the combination of palmitate with L-carnitine completely, totally normalises superoxide production in the low glucose cells and was utterly harmless in the normal glucose cells. The graph speaks against the discussion.
This normalisation of superoxide generation appears to have been too much to bear, the concept that palmitic acid might be beneficial, so they separated the palmitate from the L-carnitine and tried them separately.
“mitochondrial superoxide production increased under LG conditions with the addition of 1% palmitate while L-carnitine alone returned mitochondrial superoxide levels to those similar to the NG condition (Supplemental Figure IIIb)”
Now, this sticks in my craw. This is wrong. Completely.
Here is Supplemental figure IIIb.

Any reading with an * above it is higher than the normal glucose (NG) reading. This includes the low glucose plus L-carnitine. The low glucose with carnitine value is greater than the normal glucose value, p < 0.05.
There is, however, a crucifix above the LG+LC. The superoxide production here is significantly less than that in the LG group with added neat palmitate, that’s the meaning of the crucifix. But because of the asterix we KNOW that the superoxide here is still higher than the normal glucose value, p < 0.05.
Stating that
“L-carnitine alone returned mitochondrial superoxide levels to those similar to the NG condition”
would require that you (could) remove the asterisk from the low glucose plus carnitine column.
You can't.
You might just as well say the normal glucose plus palmitate (NG+palm) is "similar to the NG condition". Which they don't. Asterisk.
Sigh. End rant.
There are many other things which are both good and bad about this paper. But, as a lipophile living in what sometimes feels like a sea of lipophobia, it really ticks me off to see frankly incorrect and unsupported statements like this. I still scratch my head about motive.
It’s quite clear that palmitate/L-carnitine normalised the excess superoxide production of low glucose completely. I have lots of palmitate in my blood stream, though perhaps not the 1% chemical grade palmitic acid used here! I've probably got reasonable amounts of L-carnitine in my cells too, being a moderate meat eater and highly fat adapted. But the big giggle would really have been to use ketones in the study. I have ketones. As Veech pointed out, many years ago now, ketones can completely replace the whole glucose/insulin metabolic pathway in an isolated rat myocardium. Very effectively.
The flip side is that the paper is looking for information about insulin overdose rather than simple low-normal glycaemia. My BG of 2.7mmol/l was in the presence of utterly basal insulin, markedly elevated free fatty acids and modestly elevated ketones. A diabetic on a sugar based diet having a little (or even quite big) accident with their insulin to starch balance will undoubtedly have suppressed FFAs waaaaay before they hypoed and will probably have a metabolism that can't remember what a ketone body is. The metabolic milieu of parenteral insulin overdose is radically different to that of ketogenic eating. You have to engage thought processes before deciding that any blood glucose below 5mmol/l produces graded superoxide overproduction and failure of nitric oxide signalling. It's understandable from a modern diabetologist point of view, just wrong to bury data which do not support your preconceptions.
If non-injecting (and non sulphonylurea popping) folks want to reduce their FFAs and eliminate ketones in order to avoid low-normal glucose levels by eating a few extra grams of carbs, that is absolutely fine. We can all ignore the biochemistry if we so wish, serious researchers do it all the time.
Personally, I still like to have a few ketones around. And rather a lot of free fatty acids.
Peter