Tucker and I have batted this paper, best known as the Carpentier Study, around by email in the past:
Acute enhancement of insulin secretion by FFA in humans is lost with prolonged FFA elevation
and it surfaced in my memory as no tweeted this on X:
"Considering you can use LA to quickly induce IR ... the answer is complicated."
Yes, it's complicated. Both correct and incorrect.
So here is Carpentier's graph of what happens when you use an hyperglycaemic clamp to 20mmol/l, ie the right hand side of the graph where the necessary infusion rate to achieve this concentration is illustrated:
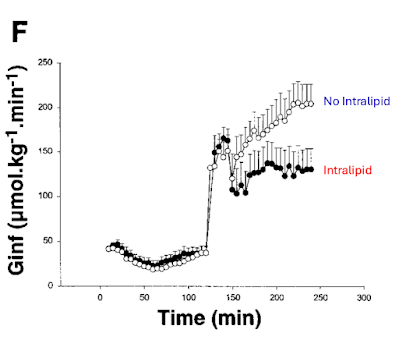
This is completely clear cut. Infusing Intralipid (~50% linoleic acid) for 48h up to and throughout the hyperglycaemic clamp markedly reduces the amount of glucose needed to maintain 20mmol/l in the blood, which signifies insulin resistance.
There are two fundamental problems here. The first is that the subjects were fed, throughout the 48h of the lead up to the clamp, a tightly controlled diet. The total number of calories is not specified but Tucker suggested from other papers by the same group that it was in the region of 2100kcal/d, designed to maintain weight stability.
This was fed either without the Intralipid or with the Intralipid, which provided an additional 1720kcal/24h, if it was included.
So in the "No Intralipid" arm the subjects were on a diet designed to maintain weight stability.
In the "Intralipid" arm the subjects were receiving 3820kcal/d, ie being calorically overloaded during the 48h leading up to the clamp.

Anyone who has even superficially glanced at
will be aware that caloric overload absolutely *should* induce insulin resistance. Otherwise there would be reductive stress (too many calories entering insulin sensitive cells) leading to an excessively high delta psi and subsequent oxidative stress, ie excessive generation of reactive oxygen species.
The control situation is very different to the Intralipid situation. They are utterly different on a overall calorie supply basis, which is fundamental to the essential adaptive nature of insulin resistance.
Okay.
The second problem (or beauty, next post) is the continuation of the infusion through the hyperglycaemic clamp. In the control situation the subjects were only receiving, intravenously, glucose at the steady state of the hyperglycaemic clamp. Around 200μmol/kg/min.
In the Intralipid arm they were receiving 40ml/h of Intralipid, ie 80kcal/h in addition to the glucose at ~130μmol/kg/min. It's beyond my willpower to convert the Intralipid supply to μmol/kg/min and we don't know the weights of the subjects anyway.
But Protons says that the calories from fat should cause enough insulin resistance to limit insulin facilitated glucose ingress to cells by an amount of calories equivalent to those supplied by the fat. This will happen with Intralipid or any other lipid emulsion, non of which was used, or was available at the time.
The issue Protons has with Intralipid is that it will not cause *enough* superoxide generation, by reverse electron transfer, to adequately resist insulin by the correct amount. If I smooth out the curves from Carpentier's paper we get this:

and if I add in what I would expect an highly saturated fat infusion to produce, we would get this this:

and if I wanted to be perverse I would predict this to be the effect of adding a safflower oil infusion (70% LA) with an even higher linoleic acid content than soybean oil:

Of course this has not been done. What has been done is the Cocoa Study by Xiao (also with Carpentier as co-author) using oral rather than intravenous fats:
which again used an hyperglycaemic clamp to 20mmol/l of glucose, which gives exactly what Protons would predict:

The hypothesis that linoleic acid generates insulin resistance promptly, as a direct effect of the generation of reactive aldenhydes formed from linoleic acid in the bloodstream, is not supported by either of the Carpentier papers discussed here.
Far more plausible is the Protons hypothesis in which linoleic acid fails to generate the ROS signal and so fails to correctly limit insulin signalling.
The same ROS signal generates satiety in the brain stem. And it also limits the insulin mediated increase in the size of adipocytes. Linoleate oxidation absolutely causes insulin resistance. No doubt. Unfortunately it doesn't cause enough insulin resistance when compared to the normal physiological mix of palmitate, stearate and oleate.
"It's complicated" applies.
Peter
Luckily the lipid peroxidation hypothesis generates the same message as the Protons hypothesis, limit linoleic acid intake. Maybe it doesn't matter which is correct, excepting it's nice to have an explanation for Carpentier's work. I have to say that the simple message "Linoleate = badness", while beautifully simple, has limited explanatory power for studies like these. "It's complicated" hits the nail on the head.
Amen.
ReplyDelete"Luckily the lipid peroxidation hypothesis generates the same message as the Protons hypothesis, limit linoleic acid intake. Maybe it doesn't matter which is correct, excepting it's nice to have an explanation for Carpentier's work. I have to say that the simple message "Linoleate = badness", while beautifully simple, has limited explanatory power for studies like these. It's complicated hits the nail on the head."
I would like to know the answer, but when you wake up in the morning and decide what you're going to eat for the day, it doesn't matter.
Yep.
ReplyDelete