Another tidy up, this time related to
Fat Quality Influences the Obesogenic Effect of High Fat Diets
and the paradox of mitochondrial uncoupling in section B of Fig 4:
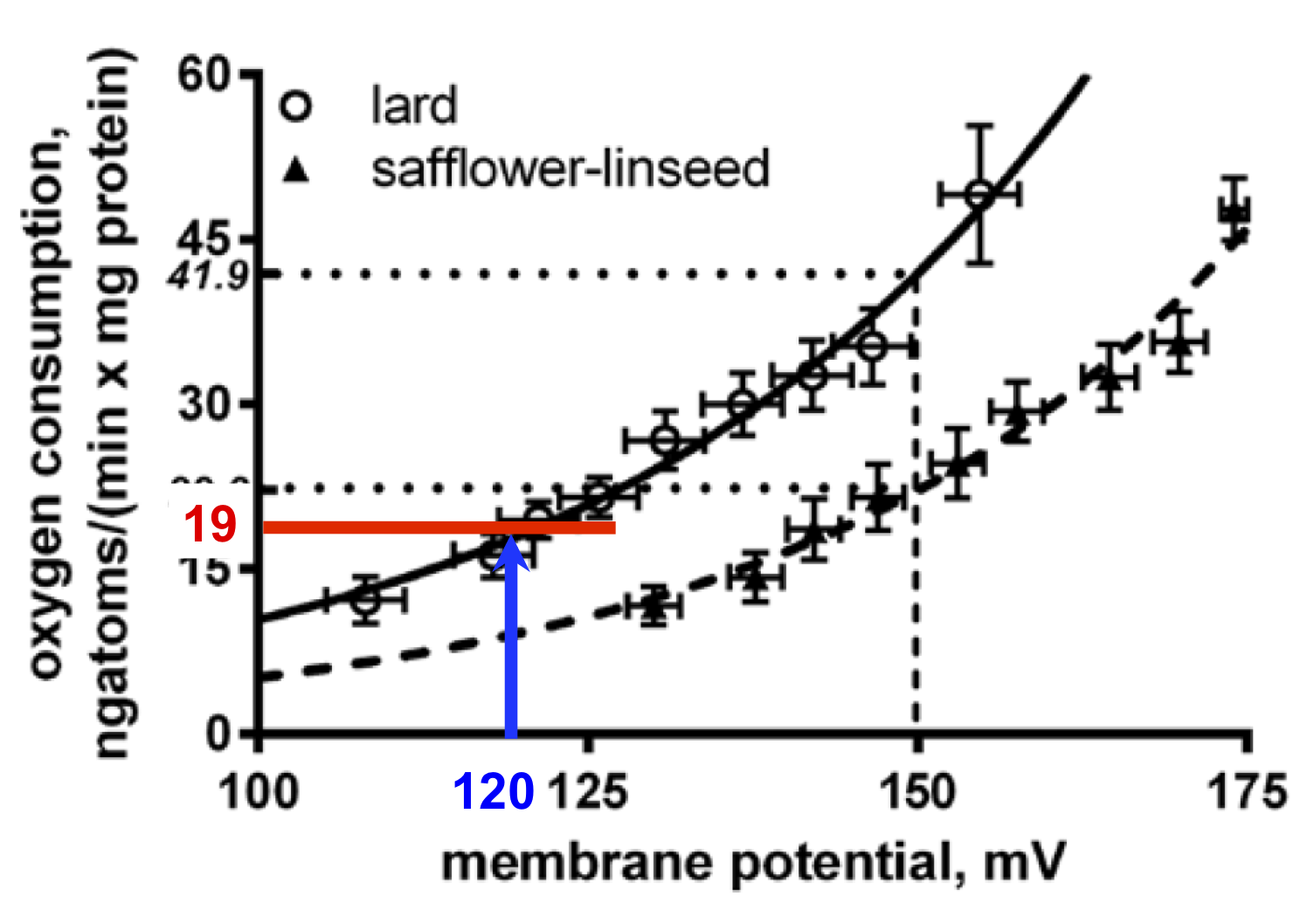
It is patently obvious from this plot that mitochondria extracted from the liver tissue of lard fed rats (consuming an obesogenic level of linoleic acid) do have an higher uncoupled oxygen consumption at all values of membrane potential when compared to the level of oxygen consumption in those rats fed the high safflower oil diet.
That is exciting and paradoxical.
We know from Figure 1 that the safflower oil fed rats were more uncoupled overall than the lard fed rats. They were synthesising much more UCP-1/cell in their brown adipose tissue and they had a greater absolute mass of brown adipose tissue by the end of the study.
They were also actively expending more energy per day at the end of the study compared to the lard fed rats. This is stated in the legend to Figure 2:
"Percent contribution of lipids, proteins and carbohydrates to total daily energy expenditure (lard = 380 ± 15, safflower-linseed = 410 ± 25 kJ/day x kg0.75) in rats fed lard or safflower-linseed high fat diet."
Safflower oil induced an initial obesity by increasing insulin sensitivity which was, by day 14, in the process of being reversed by UCP-1 reducing that excessive insulin sensitivity in WAT, assisted by activating BAT.
No one would expect hepatocytes to express UCP-1, they just don't do this. The liver deals with excess calories by sequestering them as triglycerides under the influence of insulin, sequestering them as triglycerides under the influence of succinate derived from peroxisomal omega oxidation or by signalling to BAT using FGF21 as a mediator to increase UCP-1 expression so as to bulk off-load calories as heat. But not in the liver.
Soooooo.
Safflower oil (~70% linoleic acid) produces whole-body uncoupling in the rats in the current study, apparently with the exception of within liver tissue.
Hepatocytes *do* use UCPs, they definitely synthesise UCP-2 and UCP3, but not for bulk lipid oxidation. Current thinking is they are used to fine tune their inner mitochondrial membrane potential while other signals deal with bulk caloric overload.
So the paradox is that lard fed rats have more uncoupled mitochondria than safflower fed rats. That's what the graph at the top of the page shows, ie hepatocytes are doing the opposite of what the whole rat is doing...
They measured delta psi of isolated mitochondria with a dye (safranin O) calibrated back (through 4 layers of references) to the standard technique which gives us our best estimation (don't ask) of membrane potential. They then fed isolated mitochondria in the presence of oligomycin (to block ATP synthesis) and rotenone (to prevent RET through complex I). At this point all oxygen consumption is from uncoupling. If you add increments of malonate to progressively inhibit complex II you can progressively lower the delta psi and look at the degree of uncoupling at a given titrated delta psi.
On the face of it it looks very much as if the liver really is doing the opposite to the rest of the body, which seems counter intuitive:
The degree of uncoupling is being assessed at a fixed potential, here the group chose to use 150mV (the blue line) for their example, giving an uncoupled oxygen consumption of 41.9 in lard fed vs 22.2ngatoms/(min x mg protein) of oxygen if safflower oil fed.
But is this the case in vivo? The lard fed rats are chronically underfed and have lipid locked in to adipocytes by excessive insulin sensitivity so what little lipid is being released is via augmented basal lipolysis. It is completely plausible (but also completely made-up) that they might be running a membrane potential, in vivo, as low as 120mV. Like this, blue line:
At 120mV you are not going to making a lot of ATP so uncoupling would be actively disadvantageous. In this example the uncoupled oxygen consumption would be low, in the region of 19ngatoms/(min x mg protein) of oxygen, red line.
The safflower oil fed rats went through an initial hypocaloric episode during their initial weight gain phase, but now they are uncoupling in WAT which will blunt insulin signalling and release a surfeit of FFAs, enough to supply large amounts of FFAs the liver mitochondria and (in parallel) accumulate as lipid droplets to the point of cellular damage occurring.
Under this level of direct hepatic caloric excess the mitochondrial membrane potential is likely to be high. If we run another thought experiment (ie make up) a potential of 160mV, just under that 170mV threshold for marked ROS generation, this would give us an uncoupled oxygen consumption of 29ngatoms/(min x mg protein) like this:
So, if the membrane potential differs between groups in vivo, so would the level of uncoupling. It is completely plausible that (safflower) lipid overloaded mitochondria are running an high delta psi, so need more uncoupling. Mitochondrial will never have a fixed delta psi of 150mV. It is absolutely possible that, in vivo, the safflower oil fed rats had more uncoupled hepatic mitochondria compared to the lard fed rats.
I feel much more comfortable with having hepatocytes uncouple *more* with safflower oil than with lard. The whole study is bias confirming of multiple aspects of the Protons hypothesis. Things have to make sense.
I have no problem with the mitochondrial preparation the group developed here and how they have used it. It's no better/worse than any other mitochondrial preparation. What is crucial is how you interpret the data it provides you with in the light of what must be happening physiologically. Then extrapolate backwards to the most plausible in-vivo situations, with caveats.
I have my biases.
Peter
Late addendum.
The mitochondrial uncoupling curve I have been discussing was generated from mitochondria treated with FFAs to facilitate uncoupling. Of course, if you argue that the lard fed, hypocaloric rats had lower levels of FFA in the fed state in vivo (in the fasted state there is no difference in FFA level) then there would be much less uncoupling than that discussed above, emphasising the point. Without FFA supplementation, at 160mV in the lard fed rats uncoupled oxygen consumption was as low as ~5ngatoms/(min x mg protein) in section A of Figure 4. In reality it would be somewhere between this value and the FFA supplemented value. As much as any mitochondrial prep reflects reality.