Another tidy up, this time related to
Fat Quality Influences the Obesogenic Effect of High Fat Diets
and the paradox of mitochondrial uncoupling in section B of Fig 4:



It is patently obvious from this plot that mitochondria extracted from the liver tissue of lard fed rats (consuming an obesogenic level of linoleic acid) do have an higher uncoupled oxygen consumption at all values of membrane potential when compared to the level of oxygen consumption in those rats fed the high safflower oil diet.
That is exciting and paradoxical.
We know from Figure 1 that the safflower oil fed rats were more uncoupled overall than the lard fed rats. They were synthesising much more UCP-1/cell in their brown adipose tissue and they had a greater absolute mass of brown adipose tissue by the end of the study.

They were also actively expending more energy per day at the end of the study compared to the lard fed rats. This is stated in the legend to Figure 2:
"Percent contribution of lipids, proteins and carbohydrates to total daily energy expenditure (lard = 380 ± 15, safflower-linseed = 410 ± 25 kJ/day x kg0.75) in rats fed lard or safflower-linseed high fat diet."
I would expect all rats/mice fed high safflower oil diets to use this technique and so eventually normalise their weight to that of chow fed rats/mice on a long term basis, as was found (in mice) here:
Prevention of diet-induced obesity by safflower oil: insights at the levels of PPARalpha, orexin, and ghrelin gene expression of adipocytes in mice
Okay, let's summarise:
Prevention of diet-induced obesity by safflower oil: insights at the levels of PPARalpha, orexin, and ghrelin gene expression of adipocytes in mice
Okay, let's summarise:
Safflower oil induced an initial obesity by increasing insulin sensitivity which was, by day 14, in the process of being reversed by UCP-1 reducing that excessive insulin sensitivity in WAT, assisted by activating BAT.
No one would expect hepatocytes to express UCP-1, they just don't do this. The liver deals with excess calories by sequestering them as triglycerides under the influence of insulin, sequestering them as triglycerides under the influence of succinate derived from peroxisomal omega oxidation or by signalling to BAT using FGF21 as a mediator to increase UCP-1 expression so as to bulk off-load calories as heat. But not in the liver.
Soooooo.
Safflower oil (~70% linoleic acid) produces whole-body uncoupling in the rats in the current study, apparently with the exception of within liver tissue.
Hepatocytes *do* use UCPs, they definitely synthesise UCP-2 and UCP3, but not for bulk lipid oxidation. Current thinking is they are used to fine tune their inner mitochondrial membrane potential while other signals deal with bulk caloric overload.
So the paradox is that lard fed rats have more uncoupled mitochondria than safflower fed rats. That's what the graph at the top of the page shows, ie hepatocytes are doing the opposite of what the whole rat is doing...
They measured delta psi of isolated mitochondria with a dye (safranin O) calibrated back (through 4 layers of references) to the standard technique which gives us our best estimation (don't ask) of membrane potential. They then fed isolated mitochondria in the presence of oligomycin (to block ATP synthesis) and rotenone (to prevent RET through complex I). At this point all oxygen consumption is from uncoupling. If you add increments of malonate to progressively inhibit complex II you can progressively lower the delta psi and look at the degree of uncoupling at a given titrated delta psi.
On the face of it it looks very much as if the liver really is doing the opposite to the rest of the body, which seems counter intuitive:

The degree of uncoupling is being assessed at a fixed potential, here the group chose to use 150mV (the blue line) for their example, giving an uncoupled oxygen consumption of 41.9 in lard fed vs 22.2ngatoms/(min x mg protein) of oxygen if safflower oil fed.
But is this the case in vivo? The lard fed rats are chronically underfed and have lipid locked in to adipocytes by excessive insulin sensitivity so what little lipid is being released is via augmented basal lipolysis. It is completely plausible (but also completely made-up) that they might be running a membrane potential, in vivo, as low as 120mV. Like this, blue line:
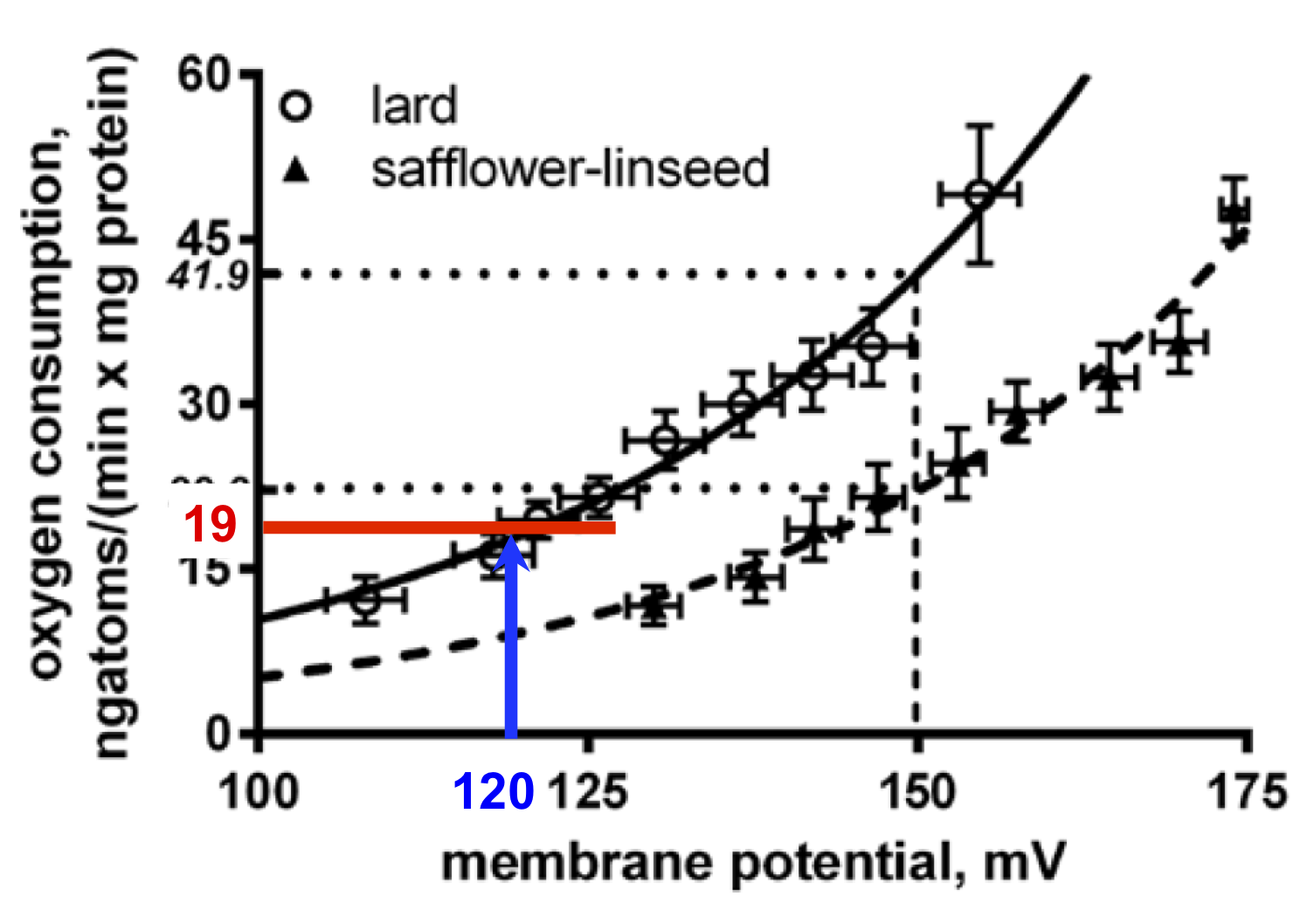
At 120mV you are not going to making a lot of ATP so uncoupling would be actively disadvantageous. In this example the uncoupled oxygen consumption would be low, in the region of 19ngatoms/(min x mg protein) of oxygen, red line.
The safflower oil fed rats went through an initial hypocaloric episode during their initial weight gain phase, but now they are uncoupling in WAT which will blunt insulin signalling and release a surfeit of FFAs, enough to supply large amounts of FFAs the liver mitochondria and (in parallel) accumulate as lipid droplets to the point of cellular damage occurring.
Under this level of direct hepatic caloric excess the mitochondrial membrane potential is likely to be high. If we run another thought experiment (ie make up) a potential of 160mV, just under that 170mV threshold for marked ROS generation, this would give us an uncoupled oxygen consumption of 29ngatoms/(min x mg protein) like this:

So, if the membrane potential differs between groups in vivo, so would the level of uncoupling. It is completely plausible that (safflower) lipid overloaded mitochondria are running an high delta psi, so need more uncoupling. Mitochondrial will never have a fixed delta psi of 150mV. It is absolutely possible that, in vivo, the safflower oil fed rats had more uncoupled hepatic mitochondria compared to the lard fed rats.
I feel much more comfortable with having hepatocytes uncouple *more* with safflower oil than with lard. The whole study is bias confirming of multiple aspects of the Protons hypothesis. Things have to make sense.
I have no problem with the mitochondrial preparation the group developed here and how they have used it. It's no better/worse than any other mitochondrial preparation. What is crucial is how you interpret the data it provides you with in the light of what must be happening physiologically. Then extrapolate backwards to the most plausible in-vivo situations, with caveats.
I have my biases.
Peter
Late addendum.
The mitochondrial uncoupling curve I have been discussing was generated from mitochondria treated with FFAs to facilitate uncoupling. Of course, if you argue that the lard fed, hypocaloric rats had lower levels of FFA in the fed state in vivo (in the fasted state there is no difference in FFA level) then there would be much less uncoupling than that discussed above, emphasising the point. Without FFA supplementation, at 160mV in the lard fed rats uncoupled oxygen consumption was as low as ~5ngatoms/(min x mg protein) in section A of Figure 4. In reality it would be somewhere between this value and the FFA supplemented value. As much as any mitochondrial prep reflects reality.

11 comments:
...some models are useful.
Another thing from the paper:
"At variance with plasma lipid profile, livers from S rats had higher lipids, triglycerides, and cholesterol, as well as higher lipid peroxidation, while water content was significantly lower, compared to L rats"
So lipid peroxidation is associated with CVD.. the lipid peroxidation was at both the systemic and heptic levels.
And if I look at the pictures of the livers, the lard fed livers appear much healthier. The elevated ALT levels hint at liver damage in the PUFA group. The PUFA group has higher heptic Trygly. PUFA fed livers weighed more. Human livers need to last a lot longer than rodents..
And this leads me back to thinking about the T2D narrative - that it is based on liver damage (with the big question about how much of the damage is reversible?).
It turns out that both high levels of LA and fructose can damage the liver - but what is particularly interesting is that the damages don't just add. This begs the question if the on going T2D pandemic is caused by the combination of high PUFA + high fructose in the average Western diet.
,.,.
Also, PUFA - with the double carbon bonds is chemically more oxidizable - so should it be a surprise that the S rats had a higher lipid oxidation rate?
Also - the change in protein oxidation could be mediated by insulin levels (which they didn't measure(why?)) - which if elevated would shut down apoptosis.
It would have added a lot if they reported insulin levels. I feel confounded by what is happening at the MT level vs hormone level at the same time - which effects dominate?
The pro PUFA narrative comes through - yet to me the health of the livers tell a quite different story?
Why are there no studies that use synthetic diets so the inputs could be restricted to a single variable at a time?
Karl,
Is T2D more closely related to pancreatic dysfunction---lack of alpha cell-beta cell cross-talk rather than liver dysfunction? Wasn't this a conclusion of Unger's work that liver is essentially normal in diabetes and was correctly responding to excess glucagon produced in pancreas.
Gyan,
I think you're correct. He thought fatty pancreas is the culprit, and glucagon excess is what underlies diabetes. In T1D, the lack of insulin results in glucagon excess. In T2D, alpha cell insulin resistance results in glucagon excess.
Unger believed that T2D is caused by lipotoxicity of islets. He discusses it in a 2014 lecture (link should be time-stamped to the relevant section).
After the rise in islet TGs, their mitochondria are almost unrecognizable, and the beta cells are degranulated. While alpha cells of healthy humans are relatively sensitive to a rise in insulin, these cells appear to be resistant to rises in insulin (i.e., glucagon levels barely go down in response to rising insulin). He blamed it on ceramides.
He also believed that in T2D, the first phase insulin response is blunted, which is critical for triggering a drop in glucagon. Even if there's still overall high levels of insulin, the insulin / glucagon ratio is low and the liver continues HGP at a good clip since it's essentially sensing a fasted state. So, yes, the liver could be working normally more or less, but receiving the wrong signals.
But, ultimately, I think Unger's model fits well with the protons perspective that LA results in excessively insulin sensitive adipocytes + insulin signaling leads to AT distension, elevated basal lipolyis, FFA leak, leading to ectopic fat accumulation, including the liver and pancreas.
Granted, Unger believed the root cause of T2D is gluttony & sloth.
Gluttony/sloth cannot be dismissed as a factor unless we regard man as an automaton.
Taubes, in my opinion, took it beyond reason when he wrote that the champion cyclists cycle BECAUSE they have excess energy..
@Gyan & Bob Kaplan
I looked at both liver and pancreas transplant papers - while it appeared that the transplants helped - it was far from definitive. It would be quite possible to do a series of targeted studies using single variable diets, and tease out what is causing the T2D pandemic. Trying to put together the story with all sorts of disconnected papers - often with dubious design begins to look like they ask the grant funders what they want the results to be - then craft a study.
It is sort of jaw dropping to realize that the T2D pandemic is actually the leading distal cause of death - and then see strange studies that first want to legitimize the advice about high PUFA diets - I suppose it will take a generation of funerals so that the science can move on?
,.,.,
Also - I find the narrow focus on insulin's effect on glucose - ignoring the effects on FFA, autophagy, etc etc etc.. to likely miss the whole picture.
I do think that the proton-switch narrative - effecting insulin sensitivity - switching between burning and growth modes - MT population can't be ignored if we want to understand how normal physiology works. That said - there is often an assumption that insulin sensitivity in different tissues is the same - while it appears that the set points of these different tissues are not at all the same.
The other bit that seems to lead thinking astray is because insulin resistance is part of the T2D syndrome - that it is a 'bad thing' - missing that only when adipose tissue is insulin resistant enough AND insulin is low enough - can people lose weight.
I'm considering the regulation of FFAs - which is more important to the body - FFA regulation or glucose regulation?
Has Gary Taubes revised his pro-vegetable oil views in his recent diabetes book?
It was always curious that the author of fat tomes on obesity would blandly say that he has not considered the effect of PUFA.
Gyan—at the risk of sending defensive of someone I admire, I asked Taubes (in article comments) a few years ago if he was looking into PUFAs or not. He was not dismissive as you imply. He said he hadn't had time to focus on that yet. From more recent comments, I think he's starting to consider it at least.
Taubes has been studying fats since long back and enough to co-author a paper in BMJ (2018) which concludes:
"Much of the evidence suggests that the risk of coronary heart disease is reduced by replacing saturated fat with polyunsaturated fats (including plant oils) but not when carbohydrate is the replacement nutrient. "
Gyan, Our cerebral cortex may be prone to gluttony and sloth but our hypothalamus is close to an automaton and it absolutely controls food intake. You can fool the cortex but not the brainstem. If people "accidentally" over consume food the brain stem is going to do something about that. Like the rats offered chow or corn oil at the same time. The corn oil is so Rewarding that they make it 80% of their calories. Do they get fat? Hahahahaha, no.
Peter
Post a Comment