This next study is pretty well supported by an established base of in-vivo and end-of-vivo studies which I think I can safely pass by and look at the induction in changes to insulin signalling, which might be interesting
Fructose Selectively Modulates c-jun N-Terminal Kinase Activity and Insulin Signaling in Rat Primary Hepatocytes
We're looking at this:
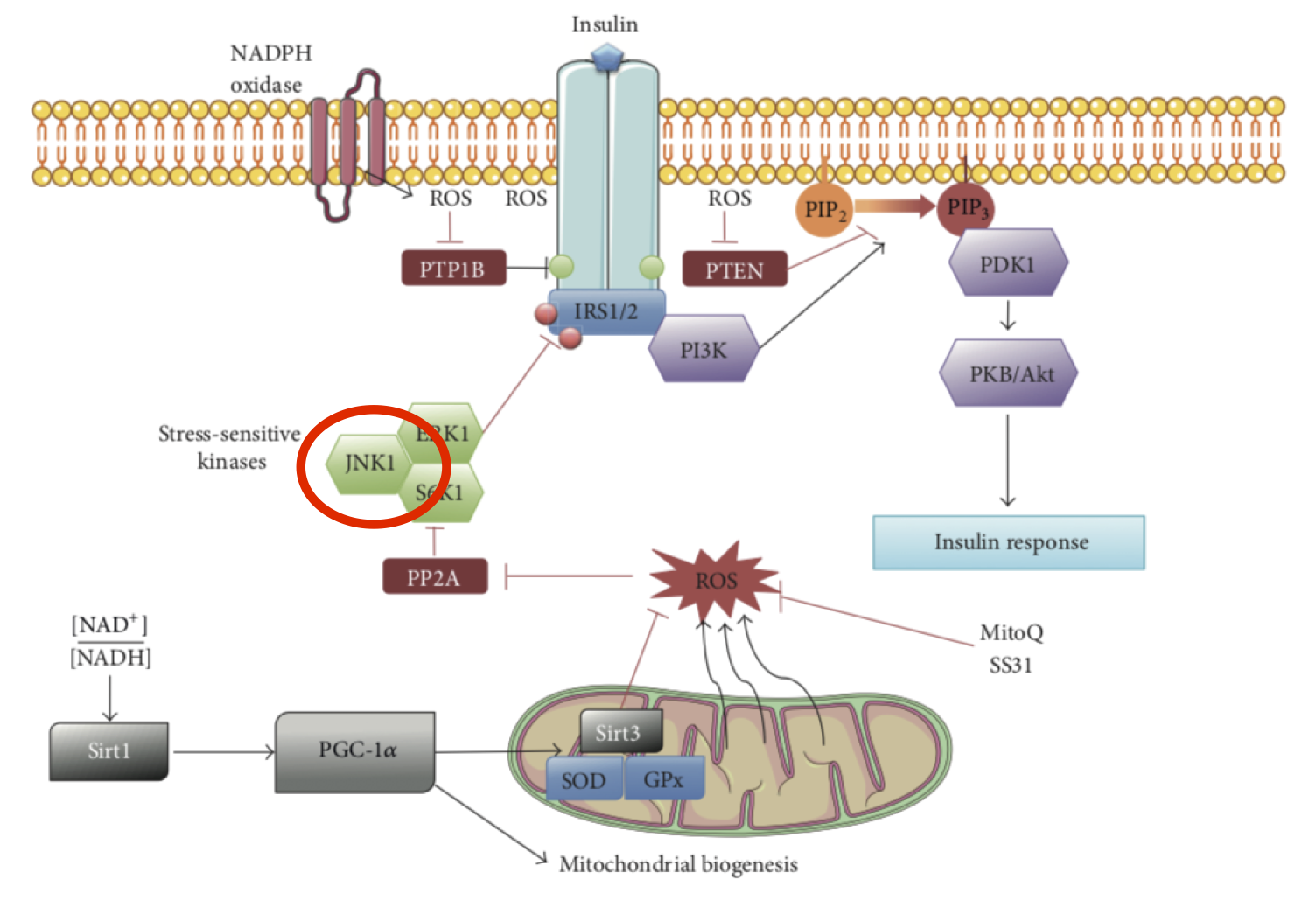
Aside: c-jun N-terminal kinase is what we expect to kill cells severely injured by being cultured in "fasting" levels of unadulterated palmitate plus glucose at 25mmol/l for 24 hours. You know the studies. Assume intolerable ROS. End aside.
The basic message from the paper is this:

The interesting parts of the paper allow us to ask hepatocytes very, very carefully, about the level of fructose exposure which inhibits insulin signalling. We can accept JNK activation as a crucial messaging step in converting fructose exposure to insulin resistance. The concept that this might be ROS driven is my own rather than anything in the paper per se.
This is the bar chart:

This model used steady state fructose exposure over four hours and is quite convincing that there is a "switch" somewhere between 0.4mmol/l and 0.6mmol/l. Our previous steady state study was that of dogs using fructose at 2.22 micromol/kg/min and this achieved a portal vein fructose of 0.1mmol/l:
Inclusion of low amounts of fructose with an intraduodenal glucose load markedly reduces postprandial hyperglycemia and hyperinsulinemia in the conscious dog
Inclusion of low amounts of fructose with an intraduodenal glucose load markedly reduces postprandial hyperglycemia and hyperinsulinemia in the conscious dog
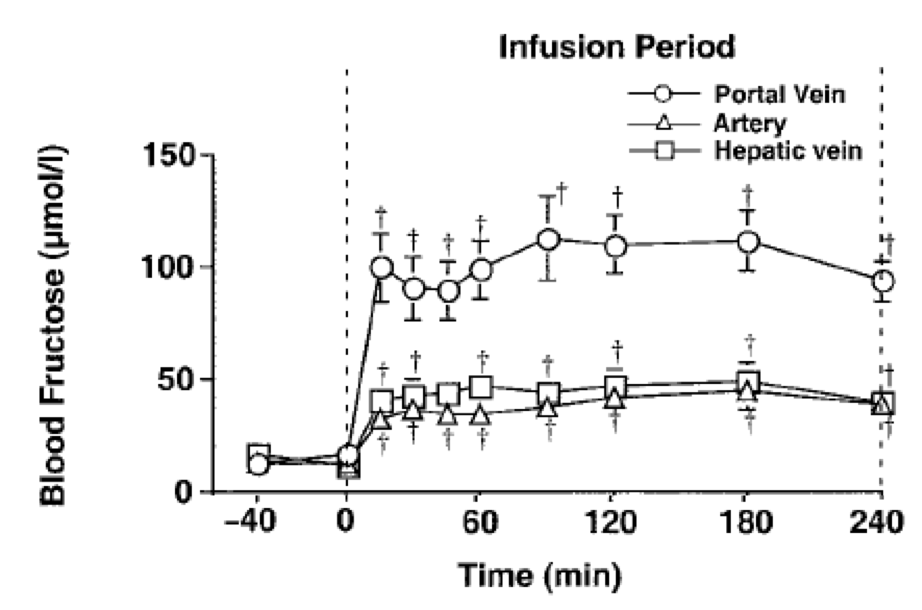
We can combine the data from both studies and suggest that, in summary, exposing hepatocytes to fructose a 0.1mmol/l is insulin signal augmenting and exposing them to fructose at 0.6mmol/l induces insulin resistance.
It is also perfectly reasonable to assume that the level of ROS which indicate that is a necessary time to induce insulin resistance are converted to a signal to be carried by the JNK pathway, exactly the one which carries the mitochondrial ROS indicator that it is necessary to induce insulin resistance.
It is also perfectly reasonable to assume that the level of ROS which indicate that is a necessary time to induce insulin resistance are converted to a signal to be carried by the JNK pathway, exactly the one which carries the mitochondrial ROS indicator that it is necessary to induce insulin resistance.
Whether the pre-emptive signal generated by palmitic acid even at low delta psi is the same one as is generated by simple caloric overload (ie failure to resist insulin in time, ie linoleic acid) remains to be seen (by me at least, so far).
We can summarise that nibbling an apple might augment storage of a bowl of porridge as hepatic glycogen but downing two cans of fructose sweetened soft drink might do other things, not least of which is to induce hepatic insulin resistance.
Looking at things fundamentally, this is a story told by ROS signalling. All the signals downstream are certainly interesting and complex but tell us little about the underlying essential process, the information derived from which they are carrying and refining.
Peter
Once again we see a feed back loop that isn't linear - there is a switch point.
ReplyDeleteOn a yet higher level - it is as if we evolved to eat an occasional fruit with some fructose, but somehow not a can of franken-food.
,.,.
I've been obsessed with switch points from back when I dug into the thyroid system. These establish homeostasis set points - such as body-weight, Blood sugar etc. I'm assuming that what sets these points is evolutionary ancient. I'm thinking hat ROS was the first loop signal for BG control - and other loops - that provide redundancy - fine tuning came later.
The mass/mess of loops in systems of biology makes teasing out reality non intuitive - especially when there is a tendency to see evolutionary design as the same as human engineering - when it is not.
,.,
OT:
Correlation in time is not proof - but this cancer trend is quite disturbing. The signal so far show no sign of regressing to the mean.
https://twitter.com/EthicalSkeptic/status/1648882315194519553?cxt=HHwWgsC-mdjugOItAAAA
OT:
ReplyDeleteThe idea that one can use a drug as a life-style-hack to get around the T2D pandemic is a risky bet. The lab mice used in safety studies have ridiculously long telemers - they are not going to show the harms of tissue damage that normal mice (or humans) would. This is backed up by a new paper:
https://www.nejm.org/doi/full/10.1056/NEJMoa2300503
Telemer length is a trade off - too long and they promote tumors - too short and early aging. (This is at odds with the popular narratives about extending life and claiming longer is better. Evolution creates what works - mess with it at your own peril).
The funny thing about the above paper is it does not reference this paper - from way back in 2001.
https://176.9.41.242/doc/longevity/2002-weinstein.pdf
I suspect that many of these mitochondrial drugs will turn out to have toxicities in the long term that in a sane world would lead to withdrawal from the market. I assume that the safety of drugs currently prescribed is way over estimated. Much better to eliminate some things from my diet than to add potential hazards.
If you have the time, Weinstein blogged into the details here:
https://odysee.com/@BretWeinstein:f/bret-and-heather-172nd-darkhorse-podcast:c
karl, I like the Weinstein paper, abstract only so far, I will try to get a listen to the podcast episode too. Obviously a 2002 paper doesn't exist in modern research parlance. The complete absence of shotgun metagenomics or metabolomics and lipidomics mean it will never get cited in the future. The fact it was probably written on papyrus and found alongside the Dead Sea Scrolls is probably another issue. I love stuff where people think things through!
ReplyDeletePeter
we saw a little fructose throughout the paleolithic, but not cans of coca-cola for years on end.
ReplyDeleteROS pass the evolutionary sniff test
Hi Peter, after reading your fructose series, i am curious!
ReplyDeleteHow do u explain the following situation:
Male, 28 years old, eating approximately 750g-1kg of sugar per day in the form of Apple and grape juice and Bananas. Additionally every day 750g of ground beef are eaten.
Result after exactly one year: no weight gain, no changes in blood markers of metabolic health, no visible increase in visceral or ectopic fat.
I know all this so detailed because this person is myself.
So serious question, how do u explain that?
Hi Peter, after reading your fructose series, i am curious!
ReplyDeleteHow do u explain the following situation:
Male, 28 years old, eating approximately 750g-1kg of sugar per day in the form of Apple and grape juice and Bananas. Additionally every day 750g of ground beef are eaten.
Result after exactly one year: no weight gain, no changes in blood markers of metabolic health, no visible increase in visceral or ectopic fat.
I know all this so detailed because this person is myself.
So serious question, how do u explain that?
Ah Basti, the series is far from over!
ReplyDeleteUltimately your answer is a derivative of this paper
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4019243/
which can be summarised as "FGF21", another very, very tricksie molecule. So then the question becomes why some fructose models induce FGF21 mediated weight loss and some models induce fatty liver, adipose dysplasia and (very mild) obesity?
If I were to suggest that the "choice" between fatty liver vs thermogenesis in BAT might be decided by the sensitivity of the liver to the lipogenic storage activity of insulin, might you start to think about linoleic acid? I would... The above study used 7% of calories as soybean oil, 3.5% linoleic acid in the intervention diets. Neither was obesogenic. In the follow-on study
https://www.sciencedirect.com/science/article/pii/S0955286317301948?via%3Dihub
they increased the LA (without saying) to around 6.5% of calories, which made the starch arm obesogenic but the FGF21 effect still dominated in the sucrose arm. But there will be an LA level where fatty liver will dominate, probably between 10-15% of calories (guessing).
Surwit has shown the same sucrose effect back in the dawn of time (plus MCT effects too) before FGF21 had even been thought of:
https://pubmed.ncbi.nlm.nih.gov/7752914/
Ethanol behaves in exactly the same way. Beer belly vs skinny alcoholic, or ethanol as a weight loss supplement in a low LA and/or keto setting setting.
Sadly the garden got away from us during the trip to Spain and I'm desperately trying to catch up, so not a lot of free time.
Peter
Surwit is confusing.
DeleteRemember brad marshall from fire in a bottle?
He recently presented evidence that lauric and myristic (sic?) acid (very high in coconut fat) actively replaces linoleic acid stored in tissue.
He went on presenting data of fat tissue composition of cultures eating almost exclusively coconuts.
.. their body fat is extremely saturated, they have high metabolic rate and are incredibly lean.
So coconut oil should be highly anti met syn and obesity.
Maybe surwit being on rats explains the controversy?
Seems like its proof im not pufa toxic
DeleteYeey
karl—re OT "Correlation in time is not proof - but this cancer trend is quite disturbing."
ReplyDeleteYou may, or may not, find this of interest: "RE IgG4 and cancer, autoimmunity" https://unglossed.substack.com/p/re-igg4-and-cancer-autoimmunity
You: "Surwit is confusing".
ReplyDeleteYep. This is where we make progress! I've not had chance to listen to Brad and Tucker's podcast but it's on the to do list.
A few things to account for: Coconut is fattening in assorted outbred rat and mouse strains. Coconut, MCT oil and caprylate/octanoate are not handled the same way metabolically, depends on the MCTs. You can undoubtedly develop models in humans where low dose (IIRC caprylate???) blunts the adaptive insulin resistance of acutely over-fed subjects. Butter behaves in a remarkably similar manner in rodents.
In Jim Johnson's control mice fed a high coconut/zero fructose diet they became obese, insulin resistant and had the longest median lifespan of all experimental groups (I would guess longest median health span too). So in his model of obesity from coconut with low (~4%) LA is different from obesity from LA at 10% w/o coconut. Possibly we are talking obesity +/- 4HNE????
Me: "It's complicated".
Peter