The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency.
We should all have a picture of the ETC looking a bit like this, omitting mtG3Pdh and ignoring supercomplex formation:

Guarás et al set up a model, a proof of principle extreme. They blocked the ETC completely at either complex III, or at cytochrome C or at complex IV. They even discussed short term oxygen deprivation as a generator of ROS, equivalent of blocking the extreme end point of the ETC. If you completely block the ETC in this way then any input through FADH2 containing enzymes to CoQ will always generate a massively reduced CoQ pool, one prerequisite for reverse electron transport (RET),
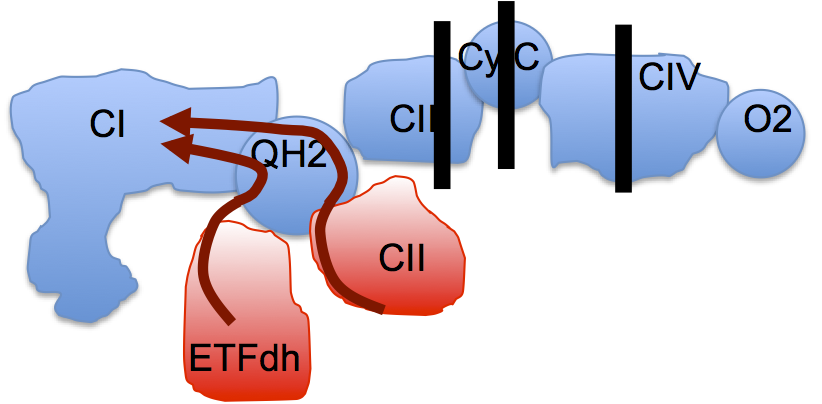
leading to the near complete disassembly of complex I. It's a model, an extreme version of reality. Fascinating in its own right, as was the ability of the fungal CoQH2 oxidase (AOX) to protect complex I completely from any of the engineered ETC defects:

AOX may not pump any protons but it does preserve complex I by reducing the extreme levels of CoQH2 which drive RET.
They subsequently went on to look at more physiological ways to generate RET and came to the conclusion that, as the balance of inputs from NADH vs FADH2 shifted, then the amount of complex I relative to complex III would need to be altered. RET is the physiological signal to balance complexes I and III against NADH and FADH2 supply.
Elegant is not the word.
They even went on to ascertain which cysteine residues were preferentially oxidised to disassemble the complex. They group around the FMN and the CoQ docking area, surprise surprise... OK. If you insist, here is the ribbon diagram from fig 5:

There's a lot of explanation in the text of what the colours and the asterisks mean.
All I really wanted to lay down with this post is that there is a physiological process where FADH2 inputs control complex I abundance. That is how it should be.
When you want a pathological model of complex I destruction, pathological levels of FADH2 input will deliver.
Peter

4 comments:
Perfect, and in tune with my LSD insight that the processes that go on in cells and mitochondria are unlikely to be just the product of Brownian motion of substrates etc between a given distribution of proteins- any mechanism that gives directionality or allows the availability of substrates to control the outputs will have been exploited.
So what (if anything) does this mean for the person with statin myopathy, whose complex 3 has been inhibited?
http://www.cell.com/cell-metabolism/pdfExtended/S1550-4131(15)00393-9
Combined with CoQ depletion... Perhaps they might increase the incidence of diabetes???????
Peter
@George
My hunch is that the myopathy is due to the statins - not the low LDL.
New Evolocumab (repatha) paper..
http://www.nejm.org/doi/full/10.1056/NEJMoa1615664#t=comments
If you look at the study - go to the comments - and yes, it looks like they are far from being forthright instead doing statistical bits to mislead.
They also say nothing about Lp(a) -- have to wonder why.
The also say nothing about oxLDL -- have to wonder why.
All cause mortality looks similar to statins..
This paper should not have been published in the form it is in.
That being said - there apparently seem to be less side effects from these PCSK9-inhibitors.
A current narrative - initial damage is done by elevated insulin. The body recruits cholesterol as part of the healing process - this healing process gets confused and starts to identify oxLDL as dying bacteria - macrophages engulf forming foam-cells that choke off artery. Thus slowing the healing process may slightly reduce heart-attack rates - but at a cost.
An alternative intervention would be to lower insulin by curing/preventing T2D - by eating lowcarb/low Linoleic acid diet. (which also reduces oxLDL ).
,.,.
So today we know that "HDL is good cholesterol" and the "lower LDL is the better" are false. That leaves trygly.
https://www.ncbi.nlm.nih.gov/labs/articles/27810046/
A lowcarb diet can easily reduce trygly. Trygly is an independent risk factor - we don't know if it is actually causative - could be that it just correlates to avoiding carbs and insulin levels.
HDL is still good - look at placebo arms in SMART or Jupiter (and no doubt FOURIER too when we get sub-group analysis). Also, look at alcohol - there's a drug that increases HDL and decreases CHD.
But factor in TG - one thing HDL is telling you is, whether there is too much TG in system, or too much insulin, as these things will also lower HDL.
Because of natural and harmless genetic variance in HDL, use fasting TG/HDL ratio for most sensitive analysis.
Clin Biochem. 2012 Jan;45(1-2):96-100. doi: 10.1016/j.clinbiochem.2011.11.001. Epub 2011 Nov 18.
The logarithm of the triglyceride/HDL-cholesterol ratio is related to the history of cardiovascular disease in patients with familial hypercholesterolemia.
Soška V, Jarkovský J, Ravčuková B, Tichý L, Fajkusová L, Freiberger T.
OBJECTIVES:
The aim of this study was to determine whether the atherogenic index of plasma (AIP=log[triglycerides/HDL-cholesterol]) differs in heterozygous familial hypercholesterolemia (FH) patients with and without a history of cardiovascular disease (CVD).
DESIGN AND METHODS:
A total of 555 FH patients with known mutations in the LDL receptor or the apolipoprotein B gene, of whom 53 had a history of CVD (CVD+ group), were retrospectively analyzed.
RESULTS:
Compared to patients without CVD (CVD- group), CVD+ patients showed significantly higher fasting LDL-cholesterol, triglycerides and AIP as well as lower HDL-cholesterol. After both adjustment for age and diabetes and using analysis based on age and sex matched groups, only the increase in triglycerides and AIP in the CVD+ vs. the CVD- group remained significant.
CONCLUSION:
The results of the present study indicate that AIP, which reflects the presence of atherogenic small LDL and small HDL particles, may be connected to the risk of CVD in FH patients.
https://www.ncbi.nlm.nih.gov/pubmed/22119890
Post a Comment