These mice were one of the core triggers to me for the concept of TCA halting due to the inability of an injured complex I to oxidise NADH. It's very clear that rotenone, at concentrations which inhibit complex I without killing your model, activates de novo lipogenesis as a technique to deal with excess NADH, excess acetyl-CoA and to provide long chain saturated fatty acids to input as FADH2 to the ETC (at ETFdh) and so to run the rest of the ETC downstream of complex I.
Look at the ability of TFAM adipocytes to uptake 2-deoxyglucose with and without insulin. This is what you get:

TFAM mouse adipocytes appear to be VERY insulin sensitive. In fact they can uptake glucose in the complete absence of any insulin, at the same rate that control (Lox) adipocytes can under supra maximal* insulin. They are insulin hypersensitive without needing any insulin...
*In the supplementary methods these adipocytes are treated with 1.25iu/kg to get this graph, but they are in cell suspension after removal and predigestion, so I don't quite see what concentration of insulin was used. I think is a reasonable assumption that the concentration would be supra maximal...
Yet these adipocytes have mitochondria with a delta psi which is profoundly depressed. The photomicrographs show them as being completely f*cked. So enhanced insulin sensitivity is completely counterintuitive.
What do they do with the glucose they so readily take up? They convert it in to lipid (supplementary data Fig2):
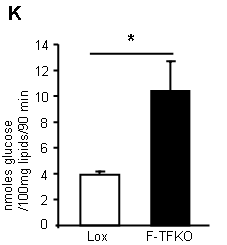
*primary isolated adipocytes, real functional cells, supplied with 5mmol/l glucose.
This lipogenesis looks remarkably like the lipogenesis you see in tissue culture cells treated with rotenone (aside; or in post-obese humans during an OGTT). These cells are energy deprived through the inability to utilise NADH. They side step this by converting acetyl-CoA to lipid and then oxidise this lipid through beta oxidation, which is less dependent on the TCA. The cells appear to be insulin sensitive because they are starving. Even without insulin there is a huge glucose uptake. Once given a few GLUT4s by supplying insulin you get supra-huge glucose uptake. I have to wonder if this is simply a concentration gradient effect, hard to say from the paper. The actual number of GLUT4s (up or down in number cf controls) is completely model dependent, if you read around. But the glucose gets converted to lipid. When you look at ATP turnover you see starvation:

*Note, cell culture 3T3-L1 derivatives at 25mmol/l glucose.
Careful here, we have gone from real mice with TFAM KO adipocytes to TFAM suppressed cell lines. Cultured shTf1 have mild knockdown, shTf2 have severe. These are tissue culture cells. They may or may not behave in quite the same manner as cells from a real live slightly broken mouse...
But the message appears to be that the processes to extract energy from glucose need to be up-regulated to the maximum possible.
Back to real broken mice. Their adipocyte mitochondria are uncoupled (i.e. they fail to correctly increase delta psi when ATP-synthase is blocked by oligomycin) and have an elevated oxygen consumption rate when running on succinate (to bypass complex I)

The uncoupling is very interesting. My feeling is that these cells really are uncoupled, really are insulin resistant due to this and that whatever effect insulin has at high doses is taken advantage of, to the maximum amount possible. Hence the term "apparently" insulin sensitive, when actually insulin resistant...
You could suggest that the underlying insulin resistance is reflected in the glycerol release. These cells, which do insulin-independent glucose uptake, also release glycerol in larger amounts than controls. Oh, and look at the palmitate oxidation too:

The FFAs released from their glycerol backbone do not appear to be discharged in to the systemic circulation (in these mice), they get oxidised because the adipocytes are in ATP starvation and can still use ETFdh to generate some ATP. We have considered FFAs and uncoupling before. The ATP situation may not suggest uncoupling as a particularly good idea but FFAs are FFAs and, if insulin is silenced, then uncoupling seems unavoidable.
BTW the control of uncoupling is so fascinating we'll have to come back to it some other time. Needless to say it integrates ADP, ATP, CoQ redox status, cytoplasm:mitochonrial ATP ratio. And free fatty acid availability, of course.
So let's summarise: Complex I damaged adipocytes are greedy for glucose without (or with) insulin. They are concurrently insulin resistant, which limits their ability to store triglycerides. Their size is immaterial. I was driven to this by a paper which demonstrated that adipocytes isolated from sucrose/lard fed mice are all equally insulin resistant, irrespective of their size (what exactly determines basal lipolysis in an interesting question, I'd guess it is not simply size, though it must be related. It's probably very important). Obviously isolated adipocytes are clear of the influence of leptin, the ventromedial hypothalamus and the sympathetic nervous system, which will certainly not be the case in any intact mouse. But, at the level of very core metabolism, this makes sense in the light of macroscopic observations. When adipocytes STOP getting fatter they do so because complex I has broken to an adequate degree. As they cease to enlarge and cease to respond to insulin they release FFAs, without any concern for higher level metabolic signalling from insulin. Systemic elevated FFAs uncouple the rest of the body's mitochondria (outside the CNS) and on we go through IGT to diabetes.
Peter

11 comments:
Peter, it looks like the superoxide in adipocytes doesn't come from mitochondria but from NADPH oxidase. Insulin activates Nox4. You'd have thought Nox4 knockouts would be fat, but they're thin, like TFAM knockouts.
http://www.jbc.org/content/287/13/10379.full
http://www.ncbi.nlm.nih.gov/pubmed/22430302
Sorry, I wrote that in a hurry and got it the wrong way round. Nox4 knockouts are fat, not thin. Nox4 makes them thin, which means it might be doing that in TFAM knockouts.
Hi Peter
I'm very uneducated when it comes to this area. So could you possibly dumb it down a little for me...ok ok a lot :) (I did say I was uneducated in this field)
I've been eating lchf for a solid 5 months now, but as far as my understanding goes, if I have high insulin won't that make my body store fat and prevent me from burning it? Even though I'm eating <10 carbs and moderate protein and very high fat?
I appreciate any response.....hopefully dumbed down to my level ;) as I know this is a old thread.
Jen x
Peter you might like to look back at this old paper.http://cancerres.aacrjournals.org/content/30/8/2223.full.pdf Hepatoma in sucrose causes swollen mitochondria. This means Ca efflux and Phosphate concentrated. Water excluded and the EZ decreases........Same thing happens in metsyn. Here is the funny thing......mitochondria also concentrate some transition metals when this happens. This means their mass rises when the swell too. When masses rises things burn fuel at a faster rate. This happens in a star that is dying and a mitochondria that is under assault. Physics 101. Burning fuels at a faster rate means more ROS and RNS. This means that the voltage on the inner mitochondrial membrane is higher when this happens. Now think about your proton series and delta psi and what the mitochondria are doing. And then remember that ketones lower the voltage when they are the primary fuel source. I think you'll get this chain of events. It begins to show you why ketosis cancer and metsyn all walk together.
Remember Robert Lustig, of the youtube video "Sugar the bitter truth"?
http://www.youtube.com/watch?v=dBnniua6-oM
I've often felt that he twisted the truth at times, maybe feeling justified because he's fighting for public health and children's in particular. But at least he's fighting the good fight and the hardcore part, where he explains very scientifically how fructose and ethanol are metabolized by the liver in the same way, that part had to be the one unquestionable fact, right?
I just watched this interesting lecture by scientist Marion Nestle.
http://www.youtube.com/watch?v=IEwRO2nyFKE
On minute 053:21 she claims she cornered Lustig and forced him to confirm if he really believes that sugar is literally a poison, as he says on his video (minute 020:25). She claims that he denied ever having said any such thing.
If you watch the 020:25 minute mark of Lustig's video you see him say that sugar is indeed a poison -which he then proceeds to repeat quite often throughout his lecture and goes at great lengths to explain the hows and the whys. That's the whole point of his now famous lecture. He didn't even say to Nestle that he's since changed his mind, he denied ever having said it in the first place.
Is there any reason why he's turning into a pathological liar? Why would he deny it when the video is evidence? Isn't the whole point of his career that sugar is poison? I just don't understand this.
Dr. Feinman raked him over the coals on this at AHS 2011.
I believe Marion Nestle honestly believes Lustig said fructose wasn't a poison.
I doubt that he actually said this--or at least that what he meant was what she understood.
He said, she said.
I wonder if DNP does the same thing to mitochondria in adipocytes?
Sorry to bring this up again, but I noticed while running DNP that weight gain seems impossible. Although I wont lose weight on DNP while still eating alot of carbs, I can absolutely pig out on junk food and not gain anything, except some water weight.
Hey guys, anyone read or have comments on this manuscript?
http://www.cell.com/cell-metabolism/abstract/S1550-4131(14)00062-X
Low Protein Intake Is Associated with a Major Reduction in IGF-1, Cancer, and Overall Mortality in the 65 and Younger but Not Older Population
These guys appear to be longevity wonks, not vegan activists. I don't know that they locked into the right cause for the association they mined from NHANES III, but seems pretty convincing that something is going on.
Might be worth a look?
--
Phillip
The age-split result was the third attempt at fracking the data, the first two attempts gave null effect for the "protein=death" hypothesis.
Third time lucky, unless there were other failed attempts they didn't write up.
It's not like the correlation was obvious from the raw data. That's always going to look suspicious.
Thanks for the info they're longevity wonks rather than vegans. I thought it was one or the other (there are so many authors it's a manifesto).
Writing about the null effects they found might have had a detrimental effect on the funding of research into a subject they obviously all feel passionate about.
Any way I thought this was interesting:
A switch in the source of ATP production and a loss in capacity to perform glycolysis are hallmarks of hepatocyte failure in advance liver disease.
http://www.ncbi.nlm.nih.gov/pubmed/24583248
In the early phase of cirrhosis, mitochondrial function and ATP generation are maintained by increasing energy production from glycolytic flux as production from oxidative phosphorylation falls. At the terminal stage of hepatic injury, mitochondria respiration and ATP production are significantly compromised, as the hepatocytes are unable to sustain the increased demand for high levels of ATP generation from glycolysis. This impairment corresponds to a decrease in glucose-6-phosphatase catalytic subunit and phosphoglucomutase 1. Similar decreased gene expression was observed in liver tissue from patients at different stages of chronic liver injury. Further, unbiased network analysis of microarray data revealed that these genes' expression was down regulated in the group of patients with poor outcome.
CONCLUSIONS:
An adaptive metabolic shift, from generating energy predominantly from oxidative phosphorylation to glycolysis, allows maintenance of energy homeostasis during early stages of liver injury, but **leads to hepatocyte dysfunction** during terminal stages of chronic liver disease because hepatocytes are unable to sustain high levels of energy production from glycolysis.
Post a Comment