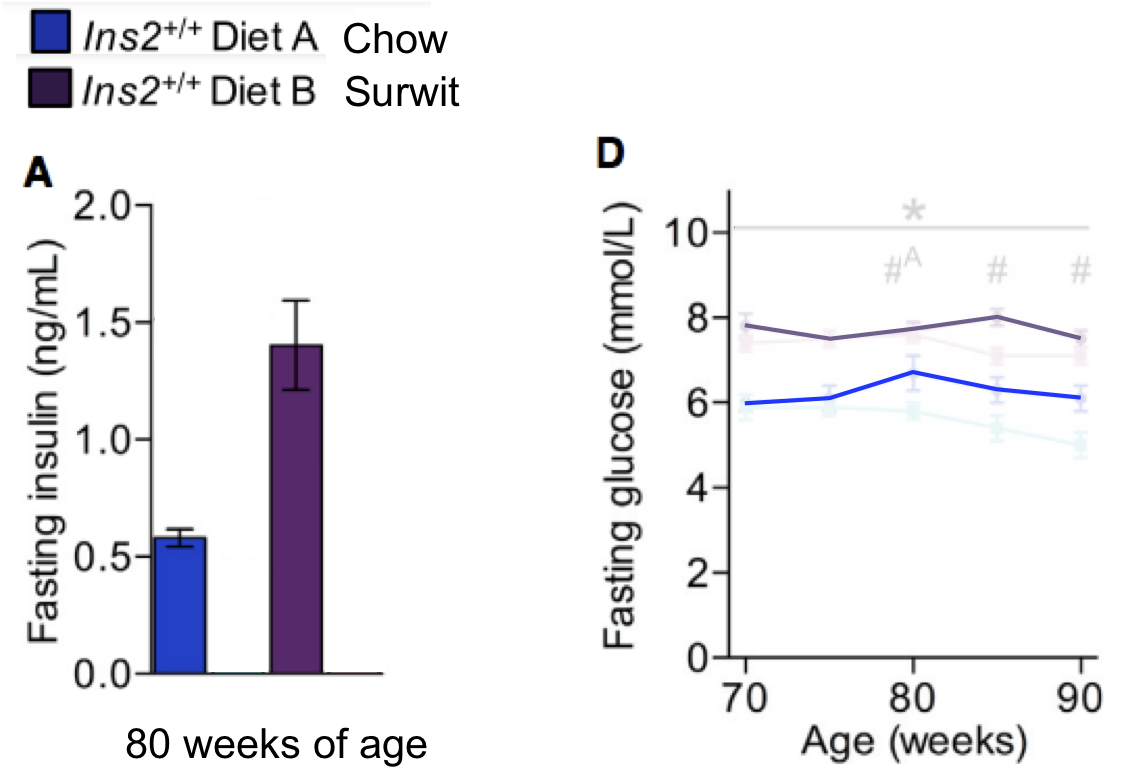
Preamble. On page 143 of Transformer (I'm assuming everyone has a copy of Transformer) Nick Lane writes:
"The deepest requirement for the proton-motive force might therefore be CO2 fixation. The prime example is the "energy converting hydrogenase" or Ech. This membrane protein has four iron-nickle-sulphur clusters, which transfer electrons from H2 to ferredoxin. Two of the clusters sit right next to a proton channel in the membrane, and their properties depend on proton binding, which is to say, the local pH. So when Ech binds protons, it can accept electrons from H2 (in the jargon, it is more easily reduced). And when the protons detach, Ech becomes more reactive, and can now force its electrons on to ferredoxin, which in turn pushes them on to CO2. Then the incoming protons bind Ech again, and the cycle repeats itself."
Which takes me back to my thoughts on pH differential driving prebiotic chemistry here:
Life (22) FeNi hydrogenase
which contains a serious logical flaw regarding the location of the site for pre-biotic carbon dioxide reduction:

In this diagram organic synthesis is happening on the acidic (pH 6), oceanic side of the FeS barrier. From where it would simply wash away in to the rest of the ocean. Metabolism actually happens on the alkaline, vent side in a constrained cell-like structure with walls made up of iron sulphide mineral.
So I've had to rethink the whole basic process with some ideas triggered by various lines in Transformer. Here we go.




The reaction which summarises the origin of life is






Structure of an Ancient Respiratory System
CO2 + H2 -> HCOOH
After formic acid formation the generation of the core building blocks of metabolism is, energetically, all down hill. I think it was Nick Lane who described this as a "free lunch you're paid to eat".
Fascinatingly, you could mix hydrogen with carbon dioxide in a jar and, even if you watched it for 4 billion years, nothing would happen. The problem is that the initial step of the conversion, which is this:
CO2 + H2 -> CO + H2O
is not a free lunch. It requires energy, it is a lack of activation energy which stops the reaction occurring spontaneously. Note it is not remotely as simple as written, the water on the right hand end just there to balance the equation, see below.
It *will* occur spontaneously if the CO2 is held at pH 6 and the H2 is held at pH10, in the presence of an iron-sulphur catalyst. An iron/nickel sulphur catalyst works even better but simple FeS seems to do the job.
So this is a crystal of iron sulphide, part of the wall of a tubule structure in a "white non-smoker" alkaline hydrothermal vent. Imagine it's a few billion atoms thick and many billions of atoms long.

In some areas of the tubule wall vent fluid is trying to pass outwards and ocean fluid is trying to pass inwards. I've depicted an interface between two flow areas as a wavy line:

To make life simpler I've now ignored the billions of other FeS layers to just show the layer adjacent to the inner side of the vent tubule. I've put in the pHs of the fluids too.

The two FeS clusters at the interface are at markedly differing pH conditions.

The red part of the intrinsically catalytic FeS surface can split a molecule of vent derived H2 in to two protons and two electrons. The protons react with hydroxyl ions in the alkaline vent fluid while the electrons hop "down hill" to the acidic FeS part of the surface in blue.

This, ephemerally, provides a negatively charged FeS surface. The electrons are clearly destined to eventually react with protons from the acidic fluid but Nick Lane explains in great detail how that process can be indirect, via CO2 interacting with the charged surface, to allow the proton and electron to recombine as an hydrogen, but this time as part of a hydrocarbon rather than as hydrogen gas. He's not particularly forthcoming on the origin of the charged surface, hence my above doodles to suggest how it might plausibly develop.
Lane goes through the conversion of CO2 to acetate at a charged surface in a step by step guide. The title of the section is "Magic surfaces" and it runs from p133 to 140 of Transformer. I've yet to find a better description. This is a wholely abiotic process. My limited summary of the "difficult" initial step is like this:

The process is laid out in Transformer, organics can be formed on the alkaline side of an FeS permeable barrier.
Nick Lane addresses the formation of this set up on page 146. He has shown that protons cross FeS barriers very easily and hydroxyl ions do so only slowly, facilitating the situation I've outline above. Nice. Electrons move over the surface of the FeS barrier briefly before combining with CO2, protons travel through the FeS barrier to provide the driving pH differential.
I've taken these concepts and overlain them on a cartoon of the power module from the membrane bound hydrogenase (MBH) of pyrococcus furiosus (I have a better cartoon for MBH than I do for Ech, they share a functionally homologous power unit so this part is interchangeable). This is Fig4 A from

which I doodled on 2019 like this:

and which I can now overlay the origin of organic synthesis scenario from my above doodles (rearranged slightly and new colour scheme) to give this

If we blank out the enzyme we can compare the two electron/proton pathways one above the other. Oh, and I've put in the Ni atom which pyrococcus uses in its hydrogenase like this:

The enzyme just needs to recapitulate the pH differential at the active site to allow the reaction to proceed. Quite how you get from this pH driven abiotic process to the same process embedded in a massive and complex enzyme system is not important here, what matters is that the basic core is clearly preserved. Getting the system to run in reverse and coupling it to an antiporter as a pump is another story.
Just in case I haven't mentioned it, the reason that the power units of Ech and MBH are so important is that they are directly homologous to the power unit of complex I and the multiple other related membrane pumps. Which means that the system, clearly derived from the inorganic process at the origin of metabolism, is pretty well ubiquitous.
The next step is to examine how a abiotic system using hydrogen as fuel can function as a proton pump which generates hydrogen as waste and what it might do with any spare electrons. That can wait for another day.
Peter