I'm just following trails from the Mrp antiporter derived subunit of the membrane bound hydrogenase of Pyrococcus furiosus. The original Mrp antiporter is, as you would hope, among those 60-odd gene families going right back to LUCA. Excellent stuff in here:
One step beyond a ribosome: The ancient anaerobic core
The final comment in the conclusions is this
"With regard to the most primitive forms of microbial physiology, microbiologists reached the same conclusion 45 years ago [26], namely that methanogens and acetogens probably represent the most ancient lineages [36]. We required 2000 genomes and powerful computers for our conclusions, while Decker et al. just thought about it. Evidently, just thinking about things can be a source of scientific progress".
That is so cool.
Peter
Thursday, February 28, 2019
Tuesday, February 26, 2019
The internet is a strange place (2)
Tom Naughton has a post up which pretty well sums up why I don't use twitter. It also encapsulates why I do respect those people who continue to do so and are willing to endure Twitter Dumb as a result of actively promoting the consumption of Food to crowds with wisdom. As opposed to the Hyperlipid approach of: "Here's the info, do what you like with it". Mea culpa.
However, I do feel Tom is a little harsh in places.
In particularly he is grossly insulting to the mental abilities of some of the more common root vegetables. If a person in category three of Twitter Dumb has insight comparable to that of a turnip, how does that make the turnip feel? More fascinatingly, how do Twitter Dumb folks survive in the real world?
Happily the explanation for the continued existence of the average category three Twitter Dumb is summarised in this paper. Most of which is composed of category three Twitter Dumb concepts
Processed foods and food reward
but it includes this gem sentence:
"All organisms must procure energy to survive, and most lack higher-order brain functions that support consciousness".
There you go.
Peter
Also relevant:
https://dilbert.com/strip/2019-02-25
https://dilbert.com/strip/2019-02-26
However, I do feel Tom is a little harsh in places.
In particularly he is grossly insulting to the mental abilities of some of the more common root vegetables. If a person in category three of Twitter Dumb has insight comparable to that of a turnip, how does that make the turnip feel? More fascinatingly, how do Twitter Dumb folks survive in the real world?
Happily the explanation for the continued existence of the average category three Twitter Dumb is summarised in this paper. Most of which is composed of category three Twitter Dumb concepts
Processed foods and food reward
but it includes this gem sentence:
"All organisms must procure energy to survive, and most lack higher-order brain functions that support consciousness".
There you go.
Peter
Also relevant:
https://dilbert.com/strip/2019-02-25
https://dilbert.com/strip/2019-02-26
Sunday, February 24, 2019
Life (22) FeNi hydrogenase
OK, more doodles. More on Yu's paper.
Back in 2015 I produced this diagram based around Nick Lane's ideas and labwork:

Please note that the inclusion of three FeS clusters in the diagram is a complete fluke. No prescience involved! I went on to concentrate on the left hand side of the diagram to give this:

I apologised at the start of the following post because the diagram is upside down by modern convention. So let me turn it the correct way up here and alter the shapes a little, not changing anything basic. Like this:

This is the basic plan for a membrane bound FeNi hydrogenase. Obviously the exact shape is a cheat. Lets look at the basic structure of a real life type 4 FeNi hydrogenase, say the one from the MBH of Pyrococcus furiosus. Which looks like this, ignoring the pumping/antiporting subunits (not shown):

Which is clear as mud. Until you overlay the doodle:

Look at those three embedded FeS clusters from the nickel catalytic core to the ferredoxin docking site, perfectly set up for electron tunneling! The H+ exit track is really as shown (though the blue arrow is my guess) and the hollow core of the gold section (MbhM) does connect to the outside. I omitted, by accident, that the track to ferredoxin is that of electrons freed from hydrogen. I'm showing the hydrogenase splitting hydrogen as per vent conditions. Nowadays it runs the other way (usually fed on fructose of all things) with hydrogen as waste.
Stuff makes sense.
Peter
Back in 2015 I produced this diagram based around Nick Lane's ideas and labwork:

Please note that the inclusion of three FeS clusters in the diagram is a complete fluke. No prescience involved! I went on to concentrate on the left hand side of the diagram to give this:

I apologised at the start of the following post because the diagram is upside down by modern convention. So let me turn it the correct way up here and alter the shapes a little, not changing anything basic. Like this:

This is the basic plan for a membrane bound FeNi hydrogenase. Obviously the exact shape is a cheat. Lets look at the basic structure of a real life type 4 FeNi hydrogenase, say the one from the MBH of Pyrococcus furiosus. Which looks like this, ignoring the pumping/antiporting subunits (not shown):

Which is clear as mud. Until you overlay the doodle:

Look at those three embedded FeS clusters from the nickel catalytic core to the ferredoxin docking site, perfectly set up for electron tunneling! The H+ exit track is really as shown (though the blue arrow is my guess) and the hollow core of the gold section (MbhM) does connect to the outside. I omitted, by accident, that the track to ferredoxin is that of electrons freed from hydrogen. I'm showing the hydrogenase splitting hydrogen as per vent conditions. Nowadays it runs the other way (usually fed on fructose of all things) with hydrogen as waste.
Stuff makes sense.
Peter
Thursday, February 21, 2019
Life (21) Pyrococcus furiosus
Pyrococcus furiosus is an interesting organism. It has a penchant for living in environments at around 100degC. It looks like it has occupied this niche for a very, very long time. It has a proton permeable, Na+ impermeable cell membrane. At 100degC constructing a proton tight membrane appears to be bloody difficult.
At some point Pyrococcus hopped from an alkaline hydrothermal vent to a volcanic black smoker type hydrothermal vent. It went, as I've argued, with a proton leaky membrane using Na+ energetics to generate ATP. Given the tools available, how did this work and what do the metabolic fossils look like?
An alkaline vent driven proto-Ech is core. It uses the proton gradient to reduce ferredoxin using molecular hydrogen. Proton (and hydroxyl) permeability is essential to neutralise the entering protons and allow the process to be continuous. At the time I wrote the Life series I felt this was unlikely to be a reversible process. I was wrong, it is.
Here is the initial proto-Ech generating reduced ferredoxin using molecular hydrogen, taken from here and here.

And here it is slightly tweaked, running in reverse, pumping protons pointlessly out through a proton permeable membrane and generating hydrogen as waste:

The second core evolutionary development is the vent proton gradient driven antiporter. This uses vent conditions to reduce the intracellular Na+ concentration:

Whatever the initial advantage of extruding Na+ from the cell might have been the major subsequent development was the formation (twice) of the Na+ ATP synthase. This Na+ ATP synthase is, ultimately, powered by the ocean to vent proton flow and permeability to protons (and hydroxyl ions) is still essential to maintain the influx of protons.
At this time life has available a proton pump, a proton leaky membrane, a proton/Na+ antiporter and a Na+ ATP synthase.
There is no point in pumping protons across a proton permeable membrane, especially if you leave the vent and every ferredoxin molecule becomes precious and life must become frugal.
What if you physically joined the proton pump and the antiporter together? So that the pumped proton stayed within the pump-antiporter complex and was never actually freed in to the environment, but was simply delivered to the proton entrance of the antiporter? So as to return to the cell, exchanging a Na+ ion outwards? Like this:
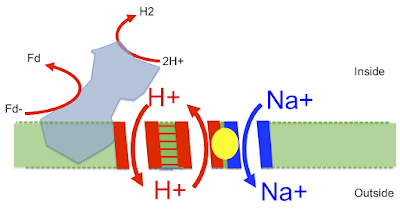
The cell membrane is still proton leaky, Na+ opaque and an Na+ ATP synthase is driven by the Na+ gradient. Like Pyrococcus furiosus today.
Does Pyrococcus have this reverse proto-Ech physically coupled to an antiporter?
Here is the image taken from this superb paper Structure of an Ancient Respiratory System:

The blue protein is what I've called proto-Ech running in reverse, generating waste H2 from reduced ferredoxin. It uses this redox reaction to pump a proton out (downwards) through the left side of the "H+ translocation module" which then (in my head) returns directly (upwards) through the right hand side of the "H+ translocation module" which is the proton half of the antiporter. This gives an associated antiported Na+ outward (downwards) via the "Na+ translocation module", second half of the antiporter. This goes off to drive Na+ energetics.
That's how it is. I think what Pyrococcus does is a derivation of exactly what LUCA would have done to leave alkaline hydrothermal vents. It's preserved due to the chance environment which makes proton tightening of the cell membrane impracticable...
Genuine Ech and Complex I are different, they use similar subunits but the redox component is on the opposite end of the intramembrane/antiporter section and this looks to be a later derivative to me, secondary to a proton tight membrane and the change to using proton pumping with blocked Na+ ingression, a far more complex process (accidental pun). And there are other Na+ pumps too. But this one in Pyrococcus, it's the one which should be there. And it is.
Made my day.
Peter
At some point Pyrococcus hopped from an alkaline hydrothermal vent to a volcanic black smoker type hydrothermal vent. It went, as I've argued, with a proton leaky membrane using Na+ energetics to generate ATP. Given the tools available, how did this work and what do the metabolic fossils look like?
An alkaline vent driven proto-Ech is core. It uses the proton gradient to reduce ferredoxin using molecular hydrogen. Proton (and hydroxyl) permeability is essential to neutralise the entering protons and allow the process to be continuous. At the time I wrote the Life series I felt this was unlikely to be a reversible process. I was wrong, it is.
Here is the initial proto-Ech generating reduced ferredoxin using molecular hydrogen, taken from here and here.

And here it is slightly tweaked, running in reverse, pumping protons pointlessly out through a proton permeable membrane and generating hydrogen as waste:

The second core evolutionary development is the vent proton gradient driven antiporter. This uses vent conditions to reduce the intracellular Na+ concentration:

Whatever the initial advantage of extruding Na+ from the cell might have been the major subsequent development was the formation (twice) of the Na+ ATP synthase. This Na+ ATP synthase is, ultimately, powered by the ocean to vent proton flow and permeability to protons (and hydroxyl ions) is still essential to maintain the influx of protons.
At this time life has available a proton pump, a proton leaky membrane, a proton/Na+ antiporter and a Na+ ATP synthase.
There is no point in pumping protons across a proton permeable membrane, especially if you leave the vent and every ferredoxin molecule becomes precious and life must become frugal.
What if you physically joined the proton pump and the antiporter together? So that the pumped proton stayed within the pump-antiporter complex and was never actually freed in to the environment, but was simply delivered to the proton entrance of the antiporter? So as to return to the cell, exchanging a Na+ ion outwards? Like this:

The cell membrane is still proton leaky, Na+ opaque and an Na+ ATP synthase is driven by the Na+ gradient. Like Pyrococcus furiosus today.
Does Pyrococcus have this reverse proto-Ech physically coupled to an antiporter?
Here is the image taken from this superb paper Structure of an Ancient Respiratory System:

The blue protein is what I've called proto-Ech running in reverse, generating waste H2 from reduced ferredoxin. It uses this redox reaction to pump a proton out (downwards) through the left side of the "H+ translocation module" which then (in my head) returns directly (upwards) through the right hand side of the "H+ translocation module" which is the proton half of the antiporter. This gives an associated antiported Na+ outward (downwards) via the "Na+ translocation module", second half of the antiporter. This goes off to drive Na+ energetics.
That's how it is. I think what Pyrococcus does is a derivation of exactly what LUCA would have done to leave alkaline hydrothermal vents. It's preserved due to the chance environment which makes proton tightening of the cell membrane impracticable...
Genuine Ech and Complex I are different, they use similar subunits but the redox component is on the opposite end of the intramembrane/antiporter section and this looks to be a later derivative to me, secondary to a proton tight membrane and the change to using proton pumping with blocked Na+ ingression, a far more complex process (accidental pun). And there are other Na+ pumps too. But this one in Pyrococcus, it's the one which should be there. And it is.
Made my day.
Peter
Saturday, February 16, 2019
The internet is a strange place
I don't do twitter or facebook (If you are a metabolic person and have friend-requested me and I've ignored it please don't take it personally, it's just not what I do with faceache) to any extent and almost never use them for metabolic subjects. By an accident of twittard I picked up this tweet by Chris Masterjohn (via raphi and Mike Eades). It made me laugh out loud and still has me giggling occasionally:
"No! Carbohydrate restriction is the stupidest approach to fatty liver ever devised. If it “works” in any case it is almost certainly by supplying more methionine and choline, not by lowering carbs. It is impossible to make more fat from carb than you get by eating fat"
I can't help but think "chylomicron", "thoracic duct" and "physiology".
Then I giggle some more.
Peter
I guess it ranks along side of "Masterjohn", "Martha" and "RQ 0.454".
Edit on 27th Feb: Published 2 days ago, very post hoc. This is what happens to surrogates for NAFLD when people apply the “stupidest approach to fatty liver ever devised”.
Post hoc analyses of surrogate markers of non-alcoholic fatty liver disease (NAFLD) and liver fibrosis in patients with type 2 diabetes in a digitally supported continuous care intervention: an open-label, non-randomised controlled study
End edit.
"No! Carbohydrate restriction is the stupidest approach to fatty liver ever devised. If it “works” in any case it is almost certainly by supplying more methionine and choline, not by lowering carbs. It is impossible to make more fat from carb than you get by eating fat"
I can't help but think "chylomicron", "thoracic duct" and "physiology".
Then I giggle some more.
Peter
I guess it ranks along side of "Masterjohn", "Martha" and "RQ 0.454".
Edit on 27th Feb: Published 2 days ago, very post hoc. This is what happens to surrogates for NAFLD when people apply the “stupidest approach to fatty liver ever devised”.
Post hoc analyses of surrogate markers of non-alcoholic fatty liver disease (NAFLD) and liver fibrosis in patients with type 2 diabetes in a digitally supported continuous care intervention: an open-label, non-randomised controlled study
End edit.
Sunday, February 10, 2019
Cell surface oxygen consumption (3) Alternative options
Have a look at this:
Limits of aerobic metabolism in cancer cells
"To gain a better understanding of cell metabolism as a function of the growth metabolic demand we performed a back-of-the-envelope calculation focusing on the major biomass components of mammalian cells".
In these days of "shotgun metabolomics" two people appear to have sat down with one or more sheets of paper (possibly larger than an envelope, though you can get some quite large envelopes I guess) and have gone back to basic first principles. They then published in a Nature journal. I love this. I feel it rates alongside getting this image published in Cell Metabolism.
TLDR for the paper:
Glucose drops through glycolysis to lactate at a rate where ATP generation per minute massively outstrips that available from mitochondrial oxphos. In redox balance. It's fast.
Anabolism from glucose consumes pyruvate (and phosphoenolpyruvate) which then forbids the pyruvate -> lactate NAD+ regeneration step. This imposes a need to avoid or deal with a cellular NADH excess.
In the Cell surface oxygen consumption (2) post I hypothesised that the regeneration of NAD+ at the cell surface would be in direct proportion to anabolism derived from pyruvate (ie glucose/glycolysis anabolism), to maintain redox balance (ie get rid of excess NADH, cycling it back to NAD+). It is particularly a feature of highly glycolytic cancer cells.
These folks appear to be saying the same thing but looking at differing cellular techniques to avoid NADH cumulation.
There's lots of other good stuff in there too. Like the rate of mitochondrial ATP generation from HeLa cell mitochondria compared to that of normal cardiac myocyte mitochondria. The ATP production via oxphos is an order of magnitude greater in mitochondria from the cardiac myocytes.
Oh, and glutaminolysis as another NADH avoiding ploy. This is the quote:
"Glutamate can be converted to citrate via reductive carboxylation. In this pathway the NAD(P)H production by glutamate dehydrogenase is compensated by the reverse activity of the NAD(P) isocitrate dehydrogenase (Fig. 1). Glutamate can be taken from the medium or generated from glutamine by glutaminase. Interestingly, arginine and proline can be produced from glutamate with concomitant consumption of NADH (Fig. 4a). This could provide an additional mechanism for NADH turnover".
Note that the glutamate is not being oxidised, it is running a small section of the TCA backwards to generate citrate for lipid synthesis, ie anabolism. This is not glutamate turning the TCA in the normal direction toward oxaloacetate to generate ATP via NADH and oxphos, because the mitochondria of cancer cells don't seem to do oxphos very well. Somewhat Seyfried supportive.
Peter
Limits of aerobic metabolism in cancer cells
"To gain a better understanding of cell metabolism as a function of the growth metabolic demand we performed a back-of-the-envelope calculation focusing on the major biomass components of mammalian cells".
In these days of "shotgun metabolomics" two people appear to have sat down with one or more sheets of paper (possibly larger than an envelope, though you can get some quite large envelopes I guess) and have gone back to basic first principles. They then published in a Nature journal. I love this. I feel it rates alongside getting this image published in Cell Metabolism.
TLDR for the paper:
Glucose drops through glycolysis to lactate at a rate where ATP generation per minute massively outstrips that available from mitochondrial oxphos. In redox balance. It's fast.
Anabolism from glucose consumes pyruvate (and phosphoenolpyruvate) which then forbids the pyruvate -> lactate NAD+ regeneration step. This imposes a need to avoid or deal with a cellular NADH excess.
In the Cell surface oxygen consumption (2) post I hypothesised that the regeneration of NAD+ at the cell surface would be in direct proportion to anabolism derived from pyruvate (ie glucose/glycolysis anabolism), to maintain redox balance (ie get rid of excess NADH, cycling it back to NAD+). It is particularly a feature of highly glycolytic cancer cells.
These folks appear to be saying the same thing but looking at differing cellular techniques to avoid NADH cumulation.
There's lots of other good stuff in there too. Like the rate of mitochondrial ATP generation from HeLa cell mitochondria compared to that of normal cardiac myocyte mitochondria. The ATP production via oxphos is an order of magnitude greater in mitochondria from the cardiac myocytes.
Oh, and glutaminolysis as another NADH avoiding ploy. This is the quote:
"Glutamate can be converted to citrate via reductive carboxylation. In this pathway the NAD(P)H production by glutamate dehydrogenase is compensated by the reverse activity of the NAD(P) isocitrate dehydrogenase (Fig. 1). Glutamate can be taken from the medium or generated from glutamine by glutaminase. Interestingly, arginine and proline can be produced from glutamate with concomitant consumption of NADH (Fig. 4a). This could provide an additional mechanism for NADH turnover".
Note that the glutamate is not being oxidised, it is running a small section of the TCA backwards to generate citrate for lipid synthesis, ie anabolism. This is not glutamate turning the TCA in the normal direction toward oxaloacetate to generate ATP via NADH and oxphos, because the mitochondria of cancer cells don't seem to do oxphos very well. Somewhat Seyfried supportive.
Peter
Sunday, February 03, 2019
Lactate as bulk energy transport
Using RQ to track whole body substrate oxidation is pretty straight forward. An RQ of 1.0 means glucose oxidation and of 0.69 indicates fat oxidation. Mixtures come out in between. It is very simple to show that glucose is routinely converted to fatty acids because in the immediate post prandial period for any rodent fed standard low fat crapinabag the RQ becomes greater than 1.0. We would expect that during the later period when the rodent is asleep/not eating there would be a lower than expected RQ (lower than the calculated food quotient, FQ) while predominantly stored fat is oxidised. But on a high carb, very low fat diet we would expect the overall averaged RQ over 24h to be a little under 1.0, ie pretty much the same as the FQ. For an hypothetical "all glucose" diet part of the glucose diverts thus via fatty acids:
Eating: Glucose minus a little O2 -> fat RQ > 1.0
Sleeping: Fat plus lots of O2 -> CO2 + H2O RQ < 1.0
CO2/O2 = 1.0 on average over 24h.
If that 24h averaged RQ was all we had to work with we would not suspect that de-novo lipogenesis ever occurred. Nice and simple.
Much more difficult to pick up is the bulk conversion of fatty acids to glucose. This produces an unusually low RQ in the short term. But if the glucose is being produced to fuel the brain during starvation then its prompt oxidation would "correct" the unusually low RQ back upwards to a fatty acid RQ. The obvious exception was noted in a metabolically fat adapted and lactating young lady during extended fasting. She made glucose and galactose from fatty acids and gave them to her baby, rather than oxidising the sugars herself. End result was an RQ of 0.454 after just over three days of fasting with continued breast feeding.
She was making sugar out of fatty acids in bulk. She might or might not have been doing the same without lactation but in the absence of donating the sugars to her infant this would never show.
So the RQ and the FQ always average out to be the same unless something very specific is happening, ie as with Martha.
Much more difficult is to ask how do you tell whether glucose is being converted to pyruvate which then enters the mitochondria to join the TCA or whether glucose converts to lactate which is then shipped in to the mitochondria. And what if you have the absolutely crazy idea that glycolysis almost always leads to lactate and that this lactate is a transferable currency between cells? Glucose is then viewed as a one way gift from liver to tissues, to be shared out between cells/tissues as lactate.
That latter view has to use tracers to look at lactate or glucose flux. Label some lactate with carbon-13 and infuse it to steady state in the plasma of a mouse. Kill the mouse promptly and humanely and look where the C-13 atoms have ended up in glycolysis and/or TCA intermediates. Repeat the process with C-13 labelled glucose. Then glutamate. And then any other intermediary metabolite which might remotely shift bulk energy around the body.
It turns out that in starch fed mice glucose and lactate are the bulk plasma energy carriers, lactate slightly more so than glucose in the fed state and much more so in the fasted state. Certainly on a molar basis, bearing in mind that a mole of glucose has twice the carbon of a mole of lactate, which makes the situation slightly more complex. But lactate labels the TCA more strongly than glucose. Not surprisingly glucose labels glycolytic intermediates better than lactate.
Free fatty acids and ketones are a separate subject in high carbohydrate/low fat fed mice but they flux remarkably little energy, at least when fasting is limited to eight hours. Brain metabolism is also another separate subject.
TLDR:
Glucose feeds glycolysis to lactate. Most of this glycolytic lactate enters the plasma pool. Plasma lactate feeds the TCA in other cells.
Now the insightful bit from near the end of the letter:
"Among the many metabolic intermediates, why does lactate carry high flux? Lactate is redox-balanced with glucose. The rapid exchange of both tissue lactate and pyruvate with the circulation may help to equate cytosolic NAD+/NADH ratios across tissues, allowing the whole body to buffer NAD(H) disturbances in any given location. Nearly complete lactate sharing between tissues effectively decouples glycolysis and the TCA cycle in individual tissues, allowing independent tissue-specific regulation of both processes. Because almost all ATP is made in the TCA cycle, each tissue can acquire energy from the largest dietary calorie constituent (carbohydrate) without needing to carry out glycolysis. In turn, glycolytic activity can be modulated to support cell proliferation, NADPH production by the pentose phosphate pathway, brain activity, and systemic glucose homeostasis. In essence, by having glucose feed the TCA cycle via circulating lactate, the housekeeping function of ATP production is decoupled from glucose catabolism. In turn, glucose metabolism is regulated to serve more advanced objectives of the organism".
What I think this is saying is that lactate supplied to the TCA/OxPhos is for "housekeeping" ie ATP production. Glycolysis is for anabolism. Neither is absolute, but I find it an interesting point of view.
So the ultimate TLDR is:
Ox-phos = housekeeping
Glycolysis = anabolism
There is probably significant fudge-room.
Peter
Eating: Glucose minus a little O2 -> fat RQ > 1.0
Sleeping: Fat plus lots of O2 -> CO2 + H2O RQ < 1.0
CO2/O2 = 1.0 on average over 24h.
If that 24h averaged RQ was all we had to work with we would not suspect that de-novo lipogenesis ever occurred. Nice and simple.
Much more difficult to pick up is the bulk conversion of fatty acids to glucose. This produces an unusually low RQ in the short term. But if the glucose is being produced to fuel the brain during starvation then its prompt oxidation would "correct" the unusually low RQ back upwards to a fatty acid RQ. The obvious exception was noted in a metabolically fat adapted and lactating young lady during extended fasting. She made glucose and galactose from fatty acids and gave them to her baby, rather than oxidising the sugars herself. End result was an RQ of 0.454 after just over three days of fasting with continued breast feeding.
She was making sugar out of fatty acids in bulk. She might or might not have been doing the same without lactation but in the absence of donating the sugars to her infant this would never show.
So the RQ and the FQ always average out to be the same unless something very specific is happening, ie as with Martha.
Much more difficult is to ask how do you tell whether glucose is being converted to pyruvate which then enters the mitochondria to join the TCA or whether glucose converts to lactate which is then shipped in to the mitochondria. And what if you have the absolutely crazy idea that glycolysis almost always leads to lactate and that this lactate is a transferable currency between cells? Glucose is then viewed as a one way gift from liver to tissues, to be shared out between cells/tissues as lactate.
That latter view has to use tracers to look at lactate or glucose flux. Label some lactate with carbon-13 and infuse it to steady state in the plasma of a mouse. Kill the mouse promptly and humanely and look where the C-13 atoms have ended up in glycolysis and/or TCA intermediates. Repeat the process with C-13 labelled glucose. Then glutamate. And then any other intermediary metabolite which might remotely shift bulk energy around the body.
It turns out that in starch fed mice glucose and lactate are the bulk plasma energy carriers, lactate slightly more so than glucose in the fed state and much more so in the fasted state. Certainly on a molar basis, bearing in mind that a mole of glucose has twice the carbon of a mole of lactate, which makes the situation slightly more complex. But lactate labels the TCA more strongly than glucose. Not surprisingly glucose labels glycolytic intermediates better than lactate.
Free fatty acids and ketones are a separate subject in high carbohydrate/low fat fed mice but they flux remarkably little energy, at least when fasting is limited to eight hours. Brain metabolism is also another separate subject.
TLDR:
Glucose feeds glycolysis to lactate. Most of this glycolytic lactate enters the plasma pool. Plasma lactate feeds the TCA in other cells.
Now the insightful bit from near the end of the letter:
"Among the many metabolic intermediates, why does lactate carry high flux? Lactate is redox-balanced with glucose. The rapid exchange of both tissue lactate and pyruvate with the circulation may help to equate cytosolic NAD+/NADH ratios across tissues, allowing the whole body to buffer NAD(H) disturbances in any given location. Nearly complete lactate sharing between tissues effectively decouples glycolysis and the TCA cycle in individual tissues, allowing independent tissue-specific regulation of both processes. Because almost all ATP is made in the TCA cycle, each tissue can acquire energy from the largest dietary calorie constituent (carbohydrate) without needing to carry out glycolysis. In turn, glycolytic activity can be modulated to support cell proliferation, NADPH production by the pentose phosphate pathway, brain activity, and systemic glucose homeostasis. In essence, by having glucose feed the TCA cycle via circulating lactate, the housekeeping function of ATP production is decoupled from glucose catabolism. In turn, glucose metabolism is regulated to serve more advanced objectives of the organism".
What I think this is saying is that lactate supplied to the TCA/OxPhos is for "housekeeping" ie ATP production. Glycolysis is for anabolism. Neither is absolute, but I find it an interesting point of view.
So the ultimate TLDR is:
Ox-phos = housekeeping
Glycolysis = anabolism
There is probably significant fudge-room.
Peter
Saturday, February 02, 2019
Lactate: Is the astrocyte-neuron lactate shuttle scuppered?
You don't usually learn much from statements which you, personally, consider likely to be correct. Annoying statements are far more productive.
Working through Seyfried's paper
Mitochondrial Substrate-Level Phosphorylation as Energy Source for Glioblastoma: Review and Hypothesis
I came across this snippet which galled me a little:
"It is glucose and not lactate that primarily drives brain energy metabolism (Allen et al., 2005; Dienel, 2012; Nortley and Attwell, 2017), making it unlikely that lactate could serve as a major energy metabolite for neoplastic GBM cells with diminished OxPhos capacity".
Now, people will realise that the astrocyte-neuron lactate shuttle is more than a little inflammatory as a subject, to say the least. Currently it is not doing too well in the face of experimental data, which are not at all straightforward to obtain. I went to Nortley and Attwell as the most recent reference. As a rather pro-lactate shuttle sort of a person I found their straw-man setting up of the shuttle rather annoying but their data potentially convincing, though I am far from certain about this. Here is the link:
Control of brain energy supply by astrocytes
This left me wondering what more pro lactate-shuttle people might be thinking nowadays, so I went via the "see all" button to locate this commentary by Tang:
Brain activity-induced neuronal glucose uptake/glycolysis: Is the lactate shuttle not required?
which is a rather more circumspect but still accepts a decreasing probability that the lactate shuttle is in any way crucial to astrocyte-neuron energetic coupling. The silver lining was this link, used to point out that in Bl6 mice whole-brain lactate extraction from plasma is essentially zero under the reasonably normal physiological conditions studied:
Glucose feeds the TCA cycle via circulating lactate
The basic concept in the paper, that lactate is the predominant metabolic substrate for the TCA is fine to me but that the source of lactate is predominantly extracellular is very counterintuitive. But the data presented are quite convincing. So I'm interested. I think it needs a little aside before talking about the paper itself and the situation in the brain in particular, so I'll post some random thoughts before looking at the paper in more detail. The biggest down side to the paper is the authors' failure to mention Schurr, the main proponent of lactate as a redox-balanced product of glucose, a very deeply insightful and much neglected observation. But then Schurr is a serious proponent of the astrocyte-neuron lactate shuttle...
Peter
Working through Seyfried's paper
Mitochondrial Substrate-Level Phosphorylation as Energy Source for Glioblastoma: Review and Hypothesis
I came across this snippet which galled me a little:
"It is glucose and not lactate that primarily drives brain energy metabolism (Allen et al., 2005; Dienel, 2012; Nortley and Attwell, 2017), making it unlikely that lactate could serve as a major energy metabolite for neoplastic GBM cells with diminished OxPhos capacity".
Now, people will realise that the astrocyte-neuron lactate shuttle is more than a little inflammatory as a subject, to say the least. Currently it is not doing too well in the face of experimental data, which are not at all straightforward to obtain. I went to Nortley and Attwell as the most recent reference. As a rather pro-lactate shuttle sort of a person I found their straw-man setting up of the shuttle rather annoying but their data potentially convincing, though I am far from certain about this. Here is the link:
Control of brain energy supply by astrocytes
This left me wondering what more pro lactate-shuttle people might be thinking nowadays, so I went via the "see all" button to locate this commentary by Tang:
Brain activity-induced neuronal glucose uptake/glycolysis: Is the lactate shuttle not required?
which is a rather more circumspect but still accepts a decreasing probability that the lactate shuttle is in any way crucial to astrocyte-neuron energetic coupling. The silver lining was this link, used to point out that in Bl6 mice whole-brain lactate extraction from plasma is essentially zero under the reasonably normal physiological conditions studied:
Glucose feeds the TCA cycle via circulating lactate
The basic concept in the paper, that lactate is the predominant metabolic substrate for the TCA is fine to me but that the source of lactate is predominantly extracellular is very counterintuitive. But the data presented are quite convincing. So I'm interested. I think it needs a little aside before talking about the paper itself and the situation in the brain in particular, so I'll post some random thoughts before looking at the paper in more detail. The biggest down side to the paper is the authors' failure to mention Schurr, the main proponent of lactate as a redox-balanced product of glucose, a very deeply insightful and much neglected observation. But then Schurr is a serious proponent of the astrocyte-neuron lactate shuttle...
Peter
Subscribe to:
Posts (Atom)
