This is the observational study from Israel through their Pfizer vaccine roll out:
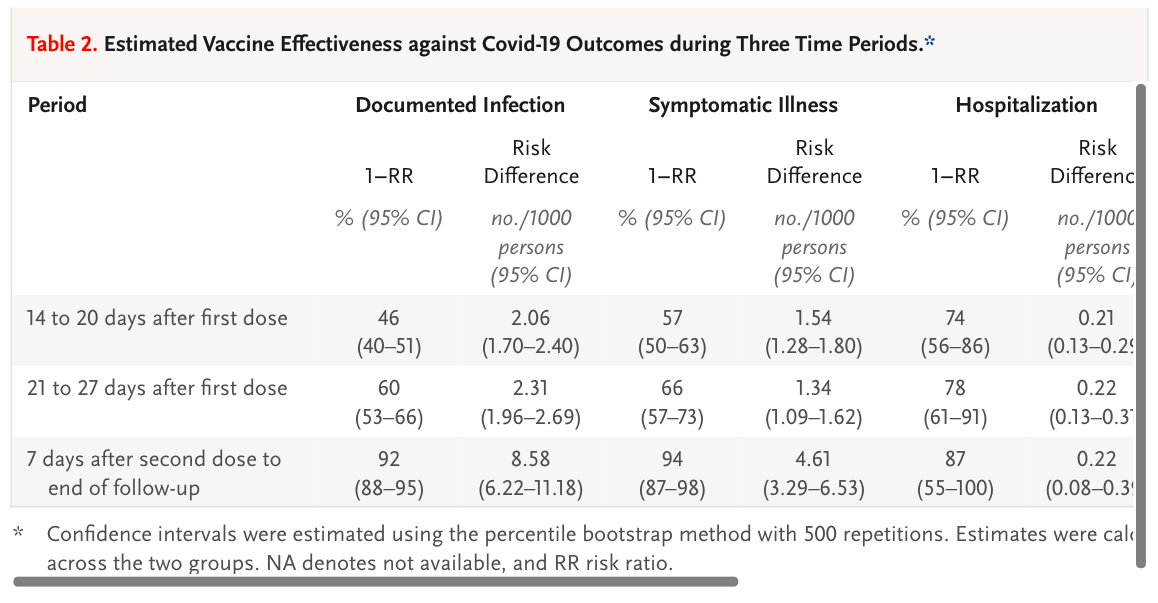
which has this excellent table showing some degree of vaccine efficacy:
Which, understandably, only starts at 14 days after first vaccination. No one would expect a vaccine to generate immunity sooner than 14 days. So we don't need to look at those initial 14 days. Interestingly, as well as excluding the first 14 days, they also excluded 26,000 care-home/housebound people and 25,000 health care workers, I've no idea why. I'd be really interested to see how the vaccine worked in elderly "crumbly" people and those who cared for them. But that's just me.
This next snippet is just a pre-print, and might stay that way. They simply reverse engineered one of the graphs in the above paper, did some basic calculations on the numbers at-risk vs infected and extracted the incidence of PCR positivity, "cases", in the earliest days of the vaccine roll out
and found, surprisingly, that people were more likely to test positive for SARS-CoV-2 in the two weeks after the vaccine compared to the non-vaccinated arm. The risk doubles. Like this:
There is a slightly sideways suggestion as to why this might occur:
"But there was a strong increase in incidence over the first week after the injection. If the increase in incidence during the first few days after immunization is a result of people being less careful after they have had their first injection, then the vaccine effectiveness after a single dose may be even greater."
ie people might have been rather less careful than they should been for the first week after the vaccine. Those Israelis are such party animals.
looking at the viral load as assessed by PCR cycle threshold, from day 1 onwards after vaccination:
These people are looking at the PCR cycle threshold value for a positive test as a surrogate for viral load, ie how badly infected a person might be. Obviously, the lower the CT value the more virus is present. It just struck me that people presenting with a requirement for PCR testing 3 days after their vaccine had twice the viral load (CT drops from 25 to 24, ie a doubling of viral load) than at any other time point. This might be random chance, but may not be.
Next we can look at the elderly people in care homes in Denmark (again just a pre-print, but less controversial this time):
Incidence went up from 488 before vaccination to 760 in the first two weeks after vaccination, before improving progressively there-after. You can see in the right hand columns for vaccine efficacy that the two weeks after vaccination show a negative efficacy value.
but this time included the AZ vector vaccine too, this will be a generic effect.
Not so easy to make out in this study, everything is expressed in Odds Ratios, ie your risk of getting infected. Non-vaccinated individuals are considered at baseline risk, ie an OR 1.0 and are highlighted with a red circle. Just being "about to be vaccinated" people (blue circle) is massively protective. I know that sounds bizarre but the baseline and pre-vaccine people will be at differing times and infection rates vary greatly over time. A falling rate of population infection (due to Gompertz/seasonality, which control everything) is massively protective against infection. Being alive after the January peak is highly protective compared to living through the December rise.

Anyhoo, we next have days 0-7 post vaccine (green arrow), higher OR of testing positive soon after your vaccine. This worsens to the blue arrowed value for days 8-20 post vaccine before the vaccine finally kicks in as shown by the yellow arrow. I'm not going to criticise the vaccine but the yellow "arrow of success" looks pretty close to the blue circle of "about to be vaccinated". Fascinating.
This is one of the first assessments of the Pfizer vaccine from way back:
Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates
and this is supplementary figure S3 section a
The Pfizer vaccine is marketed at the 30microgram dose rate, that's the brown plots. Over days 1-3 the lymphocyte count drops precipitously. If I have a patient on chemo whose lymphocytes have dropped below 1.0 10^9/l I would be worrying about immunosuppression. The effect is gone by a week. They never checked if it happens after the second dose.
Edit:
It has been pointed out to me that figure S3a is actually BNT162b1 and the vaccine actually selected for marketing was (sensibly) BNT162b2, which has a much milder effect on the lymphocyte count. So I have to consider that lymphopaenia might not be the mechanism or, if it is still the mechanism, it will only apply to those at the lower end of the box and whisker plots. The plots from BNT162b2 are here, first in 18-55 year olds:

and here for the over 55 year olds

showing a very minor effect. I still ponder whether this might have a significant effect but obviously that question is wide open. The increased infection rate is too generic to be fluke, the question is more how relevant lymphopaenia might be as an explanation.
End edit.
Lymphocytes come in many flavours. Some are irrelevant to dealing with acute infections, some are critical. On initial exposure to a virus it is the non-specific parts of the immune system which save your life in the first few days. Never mind B cells or T cells, they are critical for long term effects. In the acute phase, where the immune system has met an unknown pathogen, it's lymphocytes like natural killer cells that are going to save you. It is impossible to overstate the critical nature of the lymphocytes of the innate arm of the immune system.
To differentiate T cells from B cells from NK cells requires immunohistochemistry. This wasn't done because the lymphopaenia was considered to be transient and of no clinical significance. Bloody hell.
Lymphocytes aren't there at normal levels in the first few days after a mRNA vaccination.
To get ill you still need to meet the virus. You will still look just like a COVID-19 case, who was "probably incubating" at the time of vaccination, or went out partying under lockdown on the day you got your shot. You will still come up positive on PCR, probably with a high viral load showing as a positive result at a low cycle threshold. You might still die.
All because you were acutely immunosuppressed when you met the virus. By the vaccine. Only for a few days.
and also, browsing the FDA commentary on the early trials
Emergency Use Authorization (EUA) for an Unapproved Product Review Memorandum
I picked up this line:
"Suspected COVID-19 cases that occurred within 7 days after any vaccination were 409 in the vaccine group vs. 287 in the placebo group."
These are people who looked like they had COVID-19 soon after vaccination but did not. They were PCR tested (sometimes very repeatedly) at high PCRct and were always negative. Call them ILI, influenza like illnesses. An excess in the week after vaccination occurred. Supportive of an acute immunosuppression post vaccination. It's usually SARS-CoV-2 that gets you but any other bug can do the job too.
Does any of this matter?
Maybe.
Here in the UK we had a wave of COVID-19 sweep much of the country in the spring 0f 2020. It ended largely due to the end of the winter respiratory virus season and it failed to fully penetrate to the north of England. The virus went on vacation in the summer, as respiratory viruses do. There was a finishing off of the first wave in the autumn, mostly in the northwest then the northeast. Then it started to peter out through November.
Personally I was expecting a mild winter respiratory virus season because the gross mis-management of the situation in nursing homes earlier in the year meant that many of the people who would normally have been going to die of influenza over Christmas had already been killed in the first COVID wave and its autumn residuals. And there was minimal influenza.
This didn't happen. In early December there was a modest upswing in cases, of COVID-19 this year as it was the dominant virus at the time. Then in mid December all hell broke loose.
It didn't matter what the COVID incidence was doing at a given location; it could be stable, rising or falling, there was a surge. It was big and the shape of the surge was exactly the same in all areas. The data came and went on twitter and I never screen-shotted it. Someone recently put up a related graph of the percentage positive tests which is okay but doesn't show the spectacular shape and synchrony of the absolute numbers of positive PCR test results. Here's the picture I have, best I can find:
And here the red line marks the start of vaccination roll-out and the blue line marks the start of the surge:

Here's the graph stretched out a little to show the linear rise in positivity, in all areas, over less than a week:
I'd forgotten about these weird sudden implausible rises in infection. I'd assumed the government had changed something about the PCR protocol (which they did do in December, to catch the B117 variant which spectacularly failed to kill everyone in Kent). What rekindled my interest was India.
I think these graphs are from Ted Petrou on twitter
The rises are not as synchronous as they were in the UK last December but the shape of the curves at the right hand end of the graphs is remarkably familiar. India is huge. Vaccine roll out will not be as synchronous as it was on our tiny island off the coast of Europe. Whatever India was doing before their vaccine roll out, maybe they should have just stuck with that.
A bit of an addendum:
In the UK we largely know when people received their vaccine doses, after all they're going to be needed for vaccination passports for as long as Bojo feels like imposing them. Probably for ever.
I never, ever, expected to see what vaccination does to illness, hospitalisation and death over the first few weeks because I was pretty sure it would be appalling. It was.
So I'm utterly amazed to see this:
The study is looking at hospitalisations and deaths related to time since vaccination in a cohort being followed as part of the study. You have to be ill enough to be hospitalised to be in the study so this helpfully excludes those massive numbers of false positive PCR results being generated in December before the PCRct was finally dropped from over 40 to a more reasonable 30 (still too high). These people were ill, and they were ill with SARS-CoV-2. Genuine cases as opposed to "cases".
As the CO-CIN researchers state
"Given that the median incubation period is 5 days, the distribution indicates that most vaccinated hospitalised patients were infected around the time of vaccination, and the remainder after vaccination but before immunity had developed"
Also included is what happened to anyone's granny (A UK tier 2 category patient) if she picked up SARS-CoV-2 in the immediate aftermath of her vaccine. About 10% of these tier 2 patients didn't survive.
How could vaccination trigger infection? CO-CIN state the normal explanation:
"Elderly and vulnerable people who had been shielding, may have inadvertently been exposed and infected either through the end-to-end process of vaccination, or shortly after vaccination through behavioural changes where they wrongly assume they are immune"
Which translates as they got infected at their vaccination centre. Or these elderly people went to Israeli-style parties, during lockdown, starting on the day of their first vaccine dose. Wild things, those folks in nursing homes.
We had a winter resurgence of COVID-19. It became an epic disaster as a direct result of the immunosuppressive vaccine program being started while SARS-CoV-2 was the prevalent virus. People went through a vaccine centre where SARS-CoV-2 was persisting in aerosol form in the local environment.
No one seems to have considered that the vaccine might be directly immunosuppressive. For young fit healthy people this might not matter. In a nursing home it's a death sentence for many. They died.
Someone needs to tell the Indian government. And maybe the Japanese government too, before they cancel the Olympics.
I hope we're all enjoying being part of a global drug trial based on 40,000 fit healthy people observed for two months in mid summer. The UK government is, as in Israel, going to coerce any unwilling "volunteers" to make them take part in this on-going experiment. Thank goodness we have the Nuremberg Code, for what that is worth here.
Peter
PS, from Australia:
"Western Australia's latest cases stem from a hotel quarantine security guard in his 20s who has tested positive to COVID-19 and was potentially infectious in the community for four days.
State health authorities said the guard, who recently received his first dose of the Pfizer COVID-19 vaccine, worked on the same floor as two other positive cases from the United States and Indonesia."
No laughing. It's not funny.