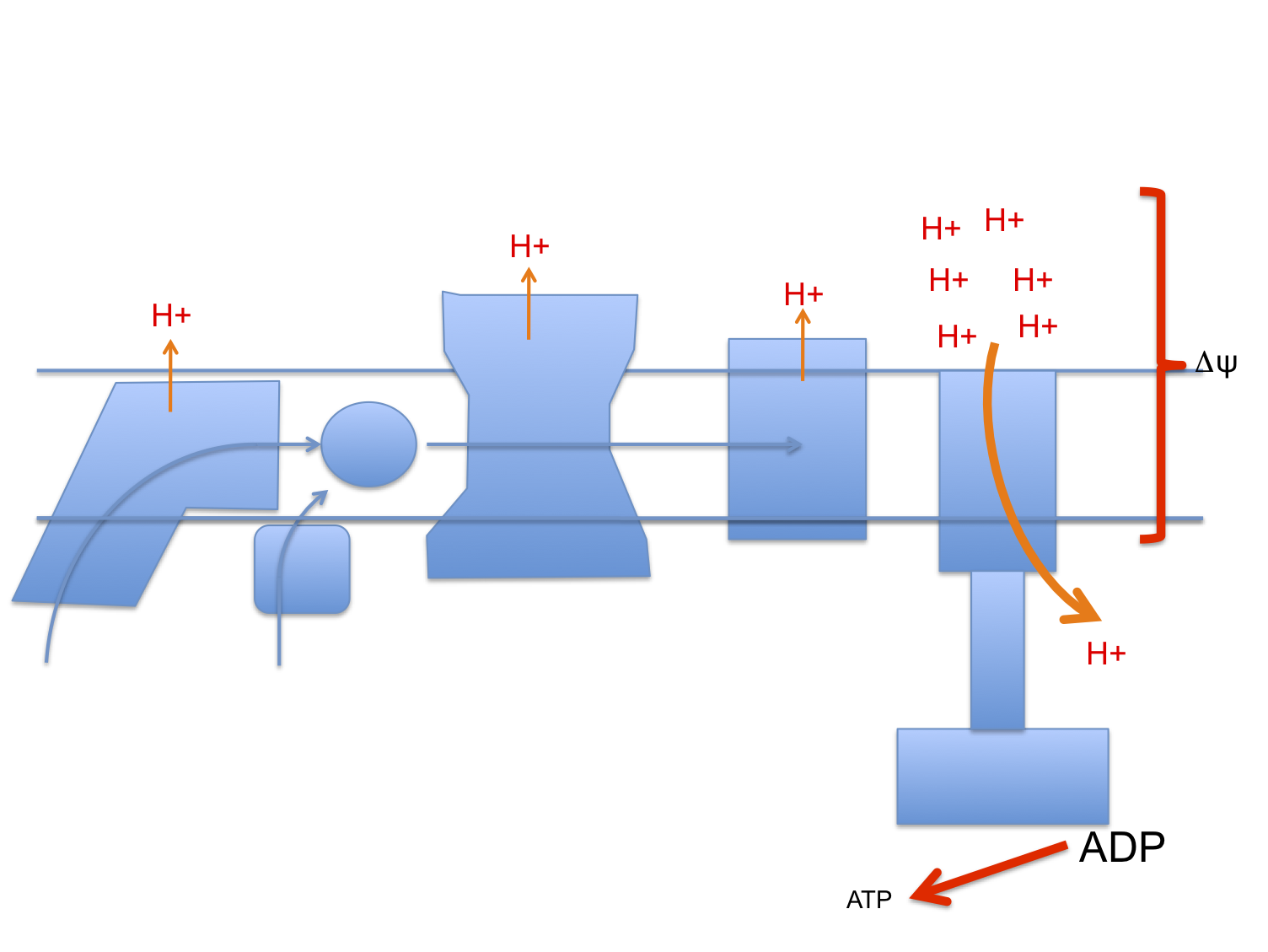
There is a considerable literature looking at "energy stress" in cells induced by marked uncoupling, usually using dinitrophenol (DNP) to profoundly reduce ATP generation from oxidative phosphorylation. The DNP concentration in cell culture to achieve this would usually be 1mM. Oral dosing can transiently achieve plasma levels in mice of around 0.5mM and is probably higher in the liver because it receives the portal blood flow from the site of absorption in the gut, so maybe this has some application to real life. Maybe. These studies are peripherally interesting as dropping ATP this aggressively triggers AMPK activation which translocates GLUT4 to the cell surface to maintain cell viability using ATP from glycolysis. The translocation is independent of any markers of insulin signalling.
An example from 1988
Evidence for two independent pathways of insulin-receptor internalization in hepatocytes and hepatoma cells
and from 2009
Regulation of glucose transporter 4 traffic by energy deprivation from mitochondrial compromise
Regulation of glucose transporter 4 traffic by energy deprivation from mitochondrial compromise
The basic premise is that ATP depletion is the activator of AMPK mediated translocation of GLT4s to the plasma membrane. However AMPK is also activated by both fasting and ketogenic diets, neither of which produces an acute ATP deficit. Years ago I suggested that a major activator of AMPK is acute loss of insulin exposure and/or its signalling, independent of ATP status.
So DNP at 1mM (1000μM) in cell culture does indeed deplete ATP and AMPK does indeed translocate GLUT4 under these circumstances. Is the mechanism of action the acute suppression of insulin signalling (secondary to the loss of mitochondrial membrane potential) rather than, or in addition to, the fall in ATP generation per se?
Happily it is quite easy to measure insulin signalling nowadays. It's also possible to use either live mice taking non-lethal doses of DNP or cell culture using therapeutic concentrations of DNP.
This next paper used DNP in live mice at non lethal dose rates and in cortical neuronal cell culture at 10-40μM concentration as opposed to 1000μM.
The Mitochondrial Uncoupler DNP Triggers Brain Cell mTOR Signaling Network Reprogramming and CREB Pathway Upregulation
Bottom line: Mild uncoupling using a therapeutic concentration of DNP suppresses insulin signalling. In this case they are looking at whole cerebral cortex in their live (until euthanasia for brain removal) mouse model or cortical neuronal cell culture.
"The protein levels of AKT, p-AKT (Thr308), ERK, and p-ERK were examined by immunoblotting which showed that the activated (phosphorylated) forms of these kinases (p-AKT and p-ERK 42/44) were reduced in the cerebral cortex at 24 and 72 h after DNP treatment (Fig. 3c–e). Collectively, these results suggest that insulin receptor signaling is suppressed in cerebral cortical cells in response to mild mitochondrial uncoupling."
"The protein levels of AKT, p-AKT (Thr308), ERK, and p-ERK were examined by immunoblotting which showed that the activated (phosphorylated) forms of these kinases (p-AKT and p-ERK 42/44) were reduced in the cerebral cortex at 24 and 72 h after DNP treatment (Fig. 3c–e). Collectively, these results suggest that insulin receptor signaling is suppressed in cerebral cortical cells in response to mild mitochondrial uncoupling."
That seriously confirms my biases.
I'm perfectly willing to extrapolate from mouse neuronal cells to mouse hepatocytes because this is a basic principle of how I view energy physiology working. I might be wrong, or not.
Low dose DNP and BAM15 will both treat metabolic syndrome in humans.
Conceptually what is happening in metabolic syndrome at the most basic level is that linoleic acid is allowing too many calories in to a cell and this leads to both storage and pathological ROS generation to side-step and/or limit the process. It is a coping mechanism for the failure of LA to signal physiological insulin resistance cf that provided by palmitate.
Uncoupling suppresses insulin signalling. If there are stored calories within the cell they are then made accessible. These calories are used for ox-phos to make up the deficit caused by the uncoupling. At normal weight (ie without excess insulin signalling due to diet) the suppression of insulin signalling will be accommodated by AMPK activation.
Stuff makes sense.
Peter