These mice were one of the core triggers to me for the concept of TCA halting due to the inability of an injured complex I to oxidise NADH. It's very clear that rotenone, at concentrations which inhibit complex I without killing your model, activates de novo lipogenesis as a technique to deal with excess NADH, excess acetyl-CoA and to provide long chain saturated fatty acids to input as FADH2 to the ETC (at ETFdh) and so to run the rest of the ETC downstream of complex I.
Look at the ability of TFAM adipocytes to uptake 2-deoxyglucose with and without insulin. This is what you get:

TFAM mouse adipocytes appear to be VERY insulin sensitive. In fact they can uptake glucose in the complete absence of any insulin, at the same rate that control (Lox) adipocytes can under supra maximal* insulin. They are insulin hypersensitive without needing any insulin...
*In the supplementary methods these adipocytes are treated with 1.25iu/kg to get this graph, but they are in cell suspension after removal and predigestion, so I don't quite see what concentration of insulin was used. I think is a reasonable assumption that the concentration would be supra maximal...
Yet these adipocytes have mitochondria with a delta psi which is profoundly depressed. The photomicrographs show them as being completely f*cked. So enhanced insulin sensitivity is completely counterintuitive.
What do they do with the glucose they so readily take up? They convert it in to lipid (supplementary data Fig2):
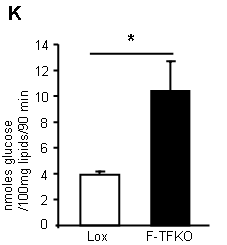
*primary isolated adipocytes, real functional cells, supplied with 5mmol/l glucose.
This lipogenesis looks remarkably like the lipogenesis you see in tissue culture cells treated with rotenone (aside; or in post-obese humans during an OGTT). These cells are energy deprived through the inability to utilise NADH. They side step this by converting acetyl-CoA to lipid and then oxidise this lipid through beta oxidation, which is less dependent on the TCA. The cells appear to be insulin sensitive because they are starving. Even without insulin there is a huge glucose uptake. Once given a few GLUT4s by supplying insulin you get supra-huge glucose uptake. I have to wonder if this is simply a concentration gradient effect, hard to say from the paper. The actual number of GLUT4s (up or down in number cf controls) is completely model dependent, if you read around. But the glucose gets converted to lipid. When you look at ATP turnover you see starvation:

*Note, cell culture 3T3-L1 derivatives at 25mmol/l glucose.
Careful here, we have gone from real mice with TFAM KO adipocytes to TFAM suppressed cell lines. Cultured shTf1 have mild knockdown, shTf2 have severe. These are tissue culture cells. They may or may not behave in quite the same manner as cells from a real live slightly broken mouse...
But the message appears to be that the processes to extract energy from glucose need to be up-regulated to the maximum possible.
Back to real broken mice. Their adipocyte mitochondria are uncoupled (i.e. they fail to correctly increase delta psi when ATP-synthase is blocked by oligomycin) and have an elevated oxygen consumption rate when running on succinate (to bypass complex I)

The uncoupling is very interesting. My feeling is that these cells really are uncoupled, really are insulin resistant due to this and that whatever effect insulin has at high doses is taken advantage of, to the maximum amount possible. Hence the term "apparently" insulin sensitive, when actually insulin resistant...
You could suggest that the underlying insulin resistance is reflected in the glycerol release. These cells, which do insulin-independent glucose uptake, also release glycerol in larger amounts than controls. Oh, and look at the palmitate oxidation too:

The FFAs released from their glycerol backbone do not appear to be discharged in to the systemic circulation (in these mice), they get oxidised because the adipocytes are in ATP starvation and can still use ETFdh to generate some ATP. We have considered FFAs and uncoupling before. The ATP situation may not suggest uncoupling as a particularly good idea but FFAs are FFAs and, if insulin is silenced, then uncoupling seems unavoidable.
BTW the control of uncoupling is so fascinating we'll have to come back to it some other time. Needless to say it integrates ADP, ATP, CoQ redox status, cytoplasm:mitochonrial ATP ratio. And free fatty acid availability, of course.
So let's summarise: Complex I damaged adipocytes are greedy for glucose without (or with) insulin. They are concurrently insulin resistant, which limits their ability to store triglycerides. Their size is immaterial. I was driven to this by a paper which demonstrated that adipocytes isolated from sucrose/lard fed mice are all equally insulin resistant, irrespective of their size (what exactly determines basal lipolysis in an interesting question, I'd guess it is not simply size, though it must be related. It's probably very important). Obviously isolated adipocytes are clear of the influence of leptin, the ventromedial hypothalamus and the sympathetic nervous system, which will certainly not be the case in any intact mouse. But, at the level of very core metabolism, this makes sense in the light of macroscopic observations. When adipocytes STOP getting fatter they do so because complex I has broken to an adequate degree. As they cease to enlarge and cease to respond to insulin they release FFAs, without any concern for higher level metabolic signalling from insulin. Systemic elevated FFAs uncouple the rest of the body's mitochondria (outside the CNS) and on we go through IGT to diabetes.
Peter
