Docosahexaenoic acid lowers cardiac mitochondrial enzyme activity by replacing linoleic acid in the phospholipidome
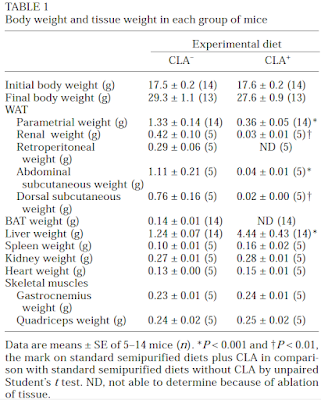
The study itself, while interesting, is not the point. The point is that the diet used contained the infamous 42% of calories from fat and was obesogenic. In common with a number of other studies I have been mulling over, the fat was butter oil. This is the butter oil composition
which has 2.3% linoleic acid in 42% of calories giving just over 1% of calories as LA. All I am interested in here is the comparison between 14 weeks on control low fat diet (CD) vs 14 weeks on a low linoleic acid Western Diet (WD). Here are the fat mass data (bodyweight mirrored this)
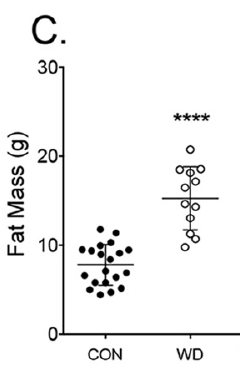
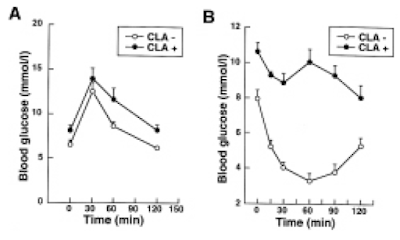
and here are the IPGTT results. We're just comparing the black control (CON) with the red western diet (WD) lines
 |
So I think we can say that 14 weeks on butter oil is obesogenic and has the appropriate insulin resistance as we expect from any obesity with its increased basal lipolysis of large adipocytes coupled with Protons effects on the non adipose tissues.
Aside:
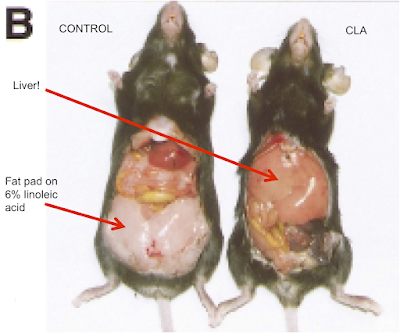
The diet looks like this:

If anyone thinks this looks like fudge, here is the recipe for a kilo of the stuff
Take 340g of table sugar, add 200g of anhydrous butter fat, bulk it up with 150g of corn starch to help it set and throw in some casein if you feel like it. Mix and extrude (you could cut in in to cubes for human consumption).
Fudge.
Looks yummy. Rewarding. Don't snigger!
End aside.
Butter oil is one of those features of study designs which produce obesity without linoleic acid.
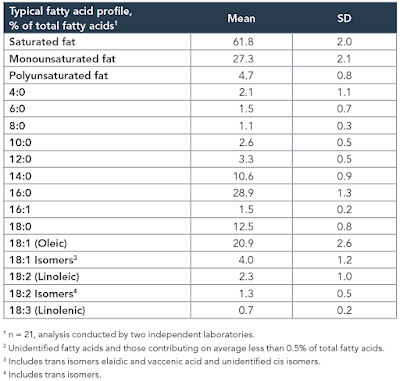
Again, prompted by Tucker, I re-drafted the F:N ratios of MUFA and PUFA in the last post to account for the consumption of one NADH to provide an NADPH for rearranging each double bond during beta oxidation and we can add these revised ratios and some for selected saturate values to the composition table of butter oil like this:
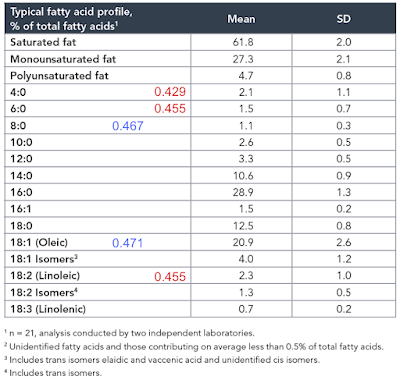
In red are linoleic acid and the short chain fatty acids of an equal or lower F:N ratio cf LA. I threw in oleic acid and C8 caprylic (blue numbers) too to point out that caprylic acid, though higher than LA, is now lower than oleic (I view oleic acid as the mammalian default for "normal" insulin sensitivity) and so might be obesogenic.
So, from the F:N ratio Protons perspective, we have a modest supply of short chain fatty acids of potentially greater insulin sensitising ability (hence obesity promoting) than linoleic acid itself.
The effect of butyrate as a dietary supplement on obesity is controversial and reviewed here
Butyrate: A Double-Edged Sword for Health?
Conclusions totally depend on how you set your study up, what you consider good and what you consider bad. Bear in mind that butyrate is the darling of fibre-philes so consider publication bias too. Conversely it is to a large extent consumed by the colonic epithelium, so not a lot gets through to the systemic circulation. But some clearly does. The snippet of Figure 2 which caught my eye was this section:
Butyrate: A Double-Edged Sword for Health?
Conclusions totally depend on how you set your study up, what you consider good and what you consider bad. Bear in mind that butyrate is the darling of fibre-philes so consider publication bias too. Conversely it is to a large extent consumed by the colonic epithelium, so not a lot gets through to the systemic circulation. But some clearly does. The snippet of Figure 2 which caught my eye was this section:
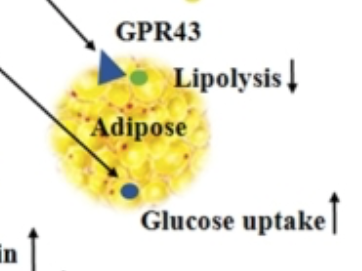
I would just point out that anything which decreases lipolysis and increases adipocyte glucose uptake is NOT going to make you skinny. It might make you insulin sensitive (bravo), at least until you get fat enough to leak excess FFAs via augmented basal lipolysis.
You could of course just say butter oil fudge is highly Rewarding, so makes rats and mice fat. How can you tell it's Rewarding? Because it makes rats and mice fat. Except corn oil is also very Rewarding but ad-lib preferential consumption fails to induce obesity. For obesity there is the absolute necessity of calories to enter adipocytes and then stay there. Long term. Dopamine release in the brain might make you choose to eat something over something else but without pathological energy storage... Shrug.
My own concept of how butter oil/sucrose causes obesity is limited by the clear fact that there is no way of simply saying a given F:N ratio will always produce obesity. Too many variables for this to be set in concrete, which allows the hypothesis to side step conflicting evidence. You have been warned.
Random thought: Sucrose (when it doesn't produce a slim insulin sensitive phenotype) usually produces hyperinsulinaemia and insulin resistance (often "skinny fat"). The higher the insulin levels the more effective insulin sensitising dietary components (linoleic acid and now possibly SCFAs) are at allowing that high level of insulin to generate obesity. Probably why the cornstarch is added to the fudge, to augment the insulin response.
As always, alternative explanations welcome.
My thanks to Basti in the comments after the last post which crystalised a lot of this current post. And to Tucker twice over.
Peter