I guess we could do worse than to being this post here;
It's a follow on study after an
initial dog study from 1998 which demonstrated that, much like glucose, in a tightly controlled somatostatin/insulin/glucagon model, low dose fructose markedly suppresses hepatic glucose output and markedly increases glycogen formation in healthy dogs. On a fixed insulin background, low dose fructose behaves as an insulin mimetic.
This follow on study in 2002 used hormonally intact dogs instrumented for portal vein, hepatic vein, femoral vein and femoral artery access. In addition they were cannulated to allow intra-duodenal infusion of glucose or glucose + fructose. This gets rid of all of the pesky delay in gastric emptying involved in an OGTT and allows time to assess effects under steady state conditions rather than the brief and dynamic changes which occur under an OGTT. Between the first and second canine studies the group had performed a couple of sets of human OGTT studies +/- fructose with somewhat challenging results, which I suspect is what led to this latest canine study, to really control as many variables as practical.
Aside: There is a horrible typo in the plasma glucose graphs throughout the study. For glucose they clearly mean milli moles/l, not micro moles/l. Arghhh. I think the fructose at 100micromoles/l is likely to be correct. I can't face trying to confirm whether their infusion rates are correct re micro vs milli. Ugh. I think their data are fine per se, unfortunate re typos. End aside.
The intra-duodenal glucose infusion was pitched to generate a modest, stable hyperglycaemia at ~10mmol/l. In the intervention section this glucose infusion remained unchanged at 44.4μmol/kg/min but an additional infusion of fructose was added at 2.2μmol/kg/min, ie around 5% fructose in addition to the original glucose. This is what happened to the systemic plasma glucose levels:
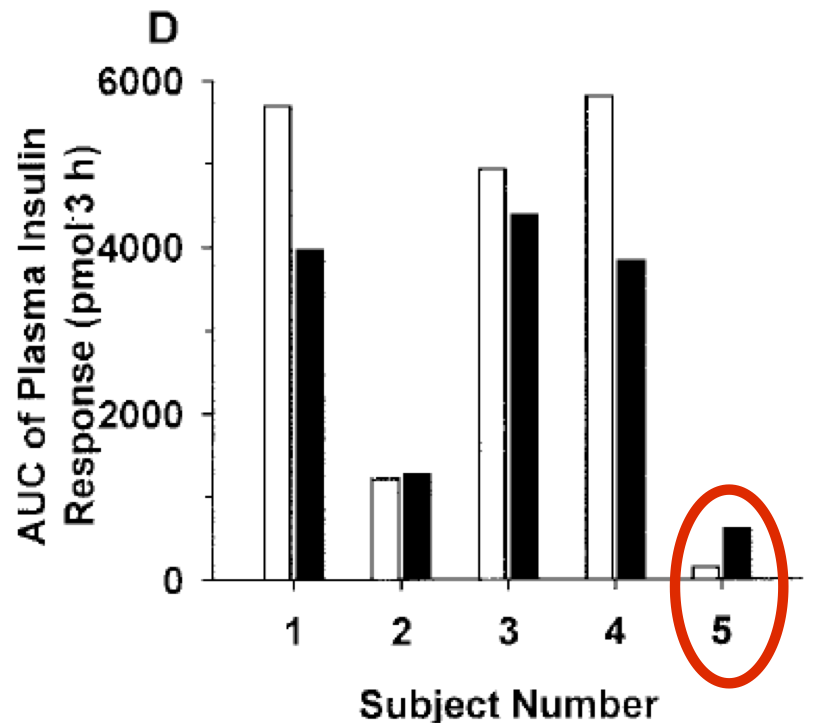
If anyone had developed a drug to produce this effect in diabetic patients it would be impressive. But that's not all. Look at what insulin levels were needed to achieve that drop in blood glucose:
Adding in 2.2μmol/kg/min of fructose to an hyperglycaemic glucose infusion profoundly lowers the insulin levels needed to maintain a modestly reduced hyperglycaemia value. In a hormonally intact dog preparation.
That's amazing if correct. Fructose appears to be profoundly insulin "sensitising" or mimicking.
Almost nothing is being "done" with the fructose. It enters the liver, drops though fructolysis and is excreted largely as lactate, well in excess of that produced by the glucose alone. I hope we're all aware that fructolysis bypasses those regulatory steps which make glycolysis a tightly regulated process. Here's the lactate change:
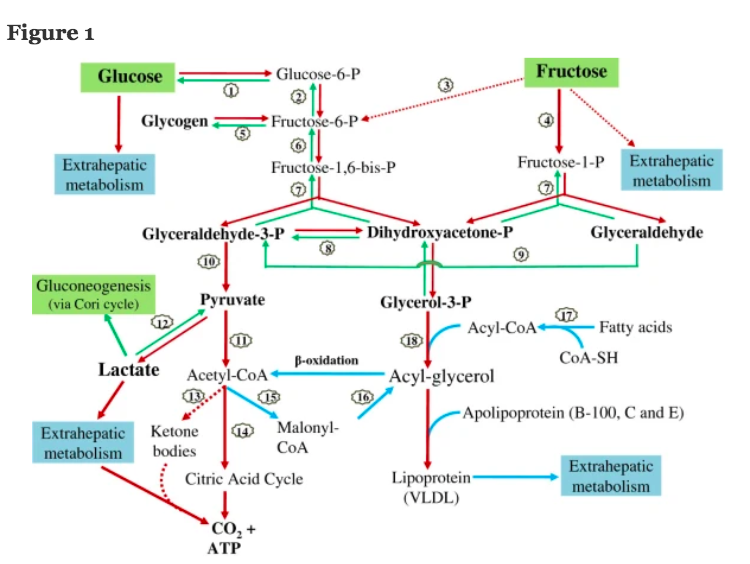
Next we can quickly doodle in the fructolysis pathway taken from here
Fructose Metabolism in Cancerwhich has this as a generic cell capable of metabolising fructose. In the liver the various GLUTs will be different but the same principle applies:
We can add in fructolysis with its unregulated, high flow pathway to lactate as typically seen in our current discussion of hepatocytes. Like this:
Equally we can put in the tightly regulated glycolysis route to either ox phos and/or lactate, usually a bit of both. But regulated by pH, citrate, ATP or oxygen availability.
We can put both pathways in together and throw in the insulin receptor too:
We are now in a position to add in the core features to an understanding of what is going on. Like this:
and then we can fade the background to let us concentrate on the generation of ROS:

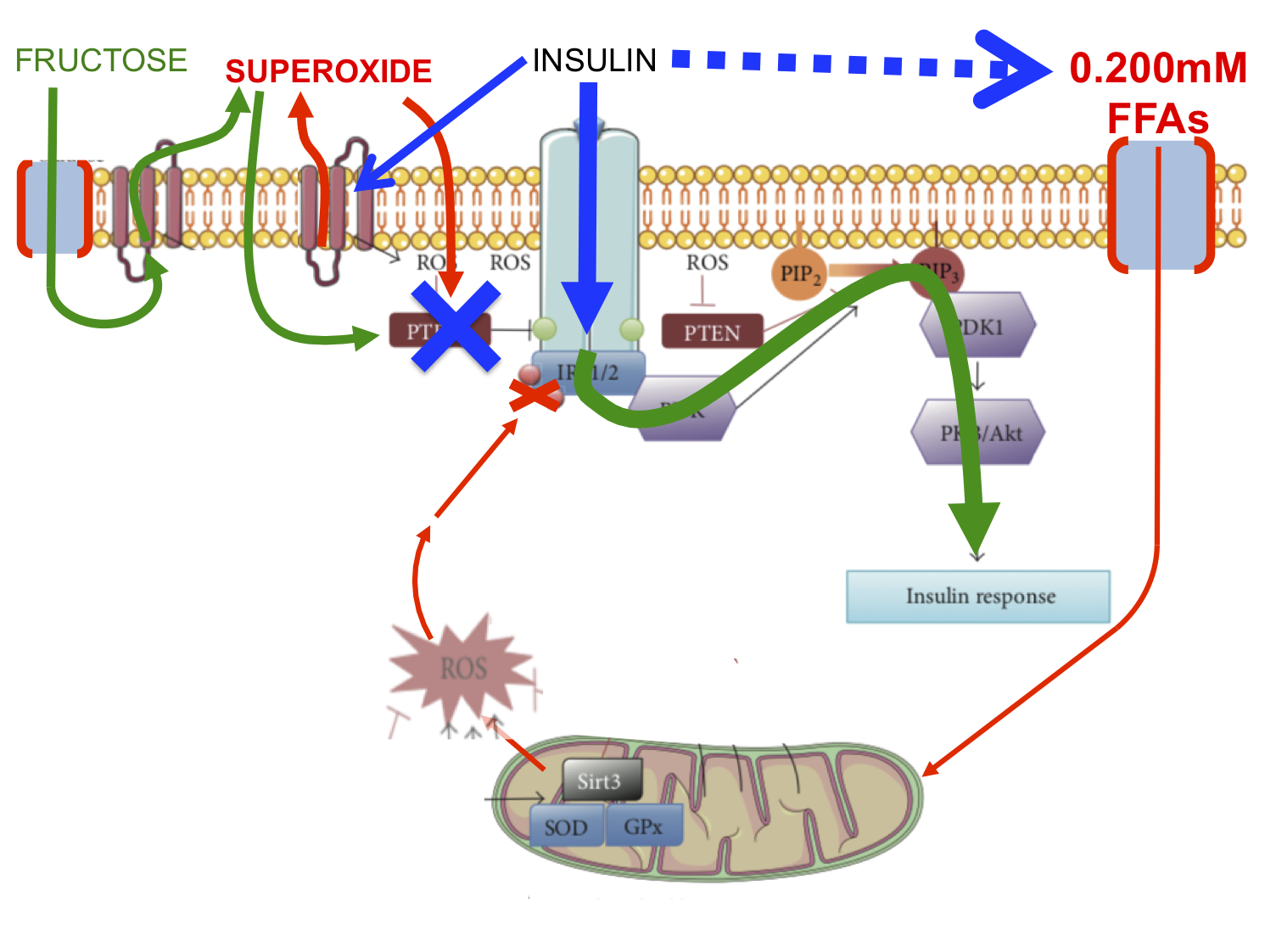
Here we have the situation under intra-duodenal glucose infusion alone. ROS are being generated by glucose ingress per se using NOX2 and also by insulin docking with the insulin receptor to activate NOX4. Both insulin level and the entrance of glucose in to cells without insulin are tightly controlled.
Neither glucose nor insulin are the signal to activate the insulin signalling cascade. That is purely the prerogative of ROS, at physiologically appropriate levels. The end result is a glucose value of around 10mmol/l and an insulin value at around 250pmol/l.
This is what happens if we add 2.2μmol/kg/min of fructose to the glucose infusion. We achieve a portal vein fructose concentration of 100μmol/l which will generate significant ROS, in direct proportion to the rate of entry of fructose to the cell, which will be high:
The fructose generated ROS will increase the ROS signal. This is the activator of the insulin cascade. The ROS signal will result in the phosphorylation of AKT, the activation of glycogen synthesis and will decrease the penetration of glucose past the liver and in to the systemic circulation. Systemic glucose will fall, less pancreatic secretion of insulin will be needed.
We get what we see in the study.
My personal definition of an obesogenic drug is one which activates "insulin" signalling in the absence of insulin. Fructose generates "unexpected" ROS when entering liver cells at a concentration of 100μmol/l. I think it is perfectly likely that, should one achieve an adipocyte exposure of 100μmol/l of fructose, that a similar effect might occur.
In Laughlin's 2014 review
there is the suggestion that peak systemic fructose concentrations healthy men after a high sugar exposure might be about 0.5mmol/l, ie 500μmol/l and a fasting level might be a fraction of a μmol/l.
There are a number of studies showing fasting fructose levels as high as 2.0mmol/l (ie 100 times Laughlin's accepted level) with post fructose loading reaching over 17.0mmol/l. That makes life easy for everyone. If you want to show fructose is harmless or beneficial you chose the low studies. If you want toxicities you choose the high studies.
But are low doses of fructose beneficial at all? Is insulin cascade activation good or bad? Are the benefits of reduced absolute insulin exposure offset by the ROS mediated activation of insulin cascade?
Whichever fructose measurement camp you fall in to, a normal human being can easily maintain systemic fructose levels > 100μmol/l in the aftermath of even a modest fructose load, which in the dog studies I consider generates enough ROS to be markedly facilitating of what we could call an insulin sensitising or mimetic exposure. Perhaps we should apply this to adipocytes.
Does fructose make you insulin sensitive? Of course it does. Does activating insulin signalling make you fat? Of course it does.
Except, of course, when it does the opposite.
Peter