This paper, which has been sitting on my hard drive for a few years, surfaced on Facebook recently. I gather it has been cited by someone who’s writing requires more ondansetron to read than I currently possess. I’ll just assume it was some sort of “eat LC and the first smidgin of myocardial hypoxia will finish you off” warning, but I’m just guessing and I have every intention of keeping it that way.
The paper itself is very convincing, well written and the protocol extensively justified. The core findings are that a LC diet impairs insulin signalling, depletes myocardial glycogen and results in massive necrosis during reperfusion after a period of myocardial hypoxia. The basic idea is that the lack of glycogen limits substrate for anaerobic glycolysis and failed insulin signalling both impairs glucose delivery from the perfusate and also fails to deliver a number of highly beneficial insulin effects which are independent of GLUT4 translocation.
This is the fate of the LC myocardium. As one of my co workers might say: Dig the hole, choose the coffin.

Obviously, for a LC eater, this is disturbing. The queue for McDougall-ism is over there.
The first thing which I find slightly disturbing is that, in a trial of the Atkins Diet™ (always mention by name), the rats ate more and were significantly heavier than the control rats, within two weeks. I’m not totally certain if I remember correctly, but I thought that the Atkins Diet was used for weight LOSS, not weight gain. Perhaps the authors might have been a little disturbed by this finding too, but apparently it doesn’t need mention. Hmmm.
The Atkins™ like diet is TestDiet 5TSY, no longer manufactured. Table I is abstracted by the authors from the full formula provided by Purina and supplies information on a need-to-know basis.
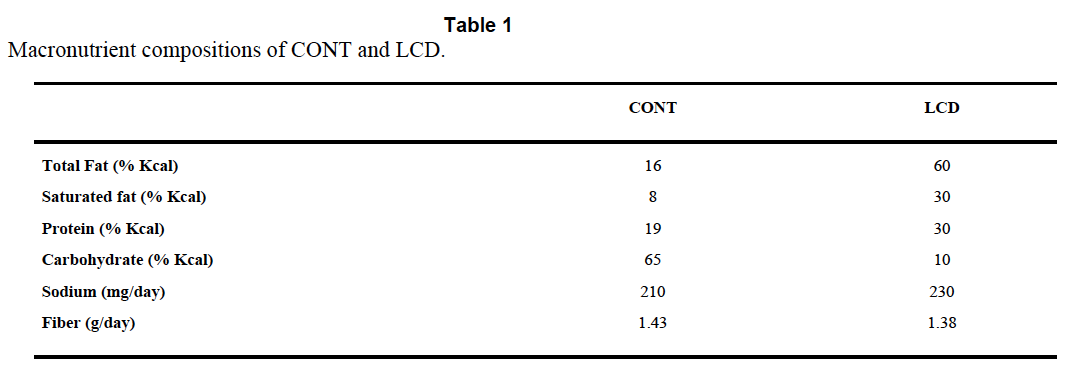
So we know total fat, saturated fat and that “The diets have the same concentrations of essential fatty acids”, i.e. we don’t quite know what the diets were made of. But Purina still will email you a pdf (very promptly) and that gives you this:

There is no mention of Crisco™ by name and no information about the trans fat content, but can you guess how much "Vegetable shortening" is in the control diet? Oh, you guessed!
I know it seems stupid to say this, but if you want to lose weight and/or survive a heart attack, for goodness sake do not choose Crisco or its equivalent as 20% of your calorie intake.
Now, this study does not tell us that a low carbohydrate diet is is good or bad for surviving a heart attack, it's simply not possible to pull that information out due to the lack of control of the variables in the diets. One can only wonder whether the formula of the Atkins Rodent Diet was specifically developed to cause metabolic problems or that somewhere along the line the original Atkins diet suggested a generous consumption of trans fat based vegetable shortening. Maybe I missed this.
I guess you could leave it there and say don't eat vegetable shortening, but there are a whole stack of follow on ideas to this study. There is MASSIVE cardiac necrosis in the LC group of rats. If it is the trans fats, how do they cause this?
One very interesting aspect of the study is the pre-ischaemia depletion of glycogen. These rats are not in ketosis. The doubling of ketone levels from 0.3mmol/l to 0.6mmol/l may well be statistically significant, but is not biologically significant. To go back, yet again, to Veech et al, we need around 5.0mmol/l of mixed ketone bodies to completely replace the insulin signalling system. You can't sidestep insulin resistance with 0.6mmol/l B-OHB. You simply cannot get rats in to a functional level of ketosis with protein at 30% of calories and carbohydrate at 12%. You need carbs near zero and protein limited to < 10% of calories to have rats in nutritional ketosis. Some papers limit protein to < 5% of calories, with minimal carbohydrate.
A period of starvation for rats on the control chow would have tested the hypothesis that it was a lack of glycogen which damaged the myocardium under hypoxia.
My own idea is that the glycogen depletion might be a surrogate for insulin resistance rather than carbohydrate restriction. We have long known that trans fatty acids from partially hydrogenated vegetable oils induce insulin resistance (though not in every study!).
So the next thought is: What sort of insulin resistance?
Are we thinking about an excess of superoxide from complex I, to imitate post prandial hyper caloric insulin resistance? Or are we thinking about the insulin resistance of fasting, when uncoupling results in blunted insulin signalling combined with high oxygen consumption and limited ATP generation?
I like the second idea when applied to this study.
If trans fatty acids, which are structurally similar but not quite identical to saturated fatty acids, allow persistent low level uncoupling outside of the physiological role of the normally structured FFAs it is interesting to speculate that they may continue to allow uncoupling when uncoupling should be utterly and totally banned.
The consequences would be decreased ATP production per unit oxygen consumed. A bit like the findings in Table 3 of this paper. The last column gives you the ATP generated per unit O2 consumed. This would be a disaster under hypoxic conditions. The IF line uses industrial trans fats. The soleus muscle is the mitochondrial dependent one cf the tibialis muscle..
ASIDE: It's worth noting that cytoplasmic ATP is a marked inhibitor of uncoupling but that this is easily overcome by adequate levels of mitochondrial ATP binding within the UCP pore from the end which does not produce a conformational change. Figure 7 gives the details but the main text is really clever stuff (without an agenda, as far as I can see). I'll blog about this paper sometime.

Under this control system the occurrence of falling mitochondrial ATP levels should allow cytoplasmic ATP to immediately shut down the normal uncoupling associated with ketogenic eating and maximise coupled ATP production per unit oxygen when this is needed. The interesting question is whether the uncoupling suspected of transfats persists under low mitochondrial ATP levels. END ASIDE.
You could speculate for hours about what trans fats may or may not do.
This is a very fertile area for idea generation. Ultimately, we don't know what a LC diet based on real Food would do to ischaemic damage in the heart. Maybe it will be as bad as a trans fat based LC (weight gain inducing) diet, maybe less severe.
We are never going to find out what Food does by using "AIN-93G Atkins/Rodent 5TSY" diet based experiments. The rats died in vain, especially as the study buried the possibility of lethal effects from trans fats.
Peter

