Mini came to us as a kitten around 2012. Back in those days I fed raw meaty bones and the only food available with bone sizes appropriate for a cat easily available in the UK was chicken wings. During bone growth I wanted to ensure adequate calcium and phosphate and nutritional grade bone meal had gone from routine use to virtually unavailable. I'd not, at that time, delved deep in to the problems of PUFA which are plentiful in the chicken wings.
Over the years she had low grade on-going gingivitis problems, with hindsight probably related to the high linoleic acid content of her diet. As I came to realise that the omega 6 PUFA are a problem I transitioned most of our other cats to 12% fat beef mince and that's what they all eat nowadays.
Not Mini. She routinely threw up on beef mince but was fine on the chicken wings, so there she stayed.
Then she broke two teeth (on a chicken wing!) and when I did her dentistry it was obvious that her low grade gingivitis was not low grade and that all of her teeth were in a bad way, so I took them all out.
No cat is going to eat raw chicken wings with her gums. I tried her on beef mince again, she threw up again. Eventually I decided to try her on chicken breast meat, well cut up and relatively low in fat so low in PUFA. She liked it, maintained weight and had no suggestion of ammonia toxicity from an almost all protein diet. She's a cat after all.
Then last November she hid up under one of the children's beds and when I managed to get her out she was very distressed and had laboured breathing. A quick trip to the practice and 30 seconds of ultrasound by someone better at it than me confirmed heart failure.
Heart failure in cats is common. It's usually hypertrophic cardiomyopathy and is essentially untreatable. I assumed that if HCM was present then high protein + high glucose (cats really do convert protein to glucose continuously) -> high GH -> IGF-1 -> cardiac hypertrophy. Probably wrong but who knows? My colleague of the ultrasound machine was uncertain if she could see hypertrophic or dilated cardiomyopathy so I booked a scan with a cardiologist. A few frusemide tablets stopped her drowning and she had no suggestion of a thrombus or pre-thrombus in either atrium.
It was dilated cardiomyopathy. Or end stage hypertrophic cardiomyopathy where replacement of cardiac myocytes with fibrous tissue looks, on ultrasound, just like the dilated form.
DCM is genetic or caused by taurine deficiency. So I PubMed-ed:
and found this:
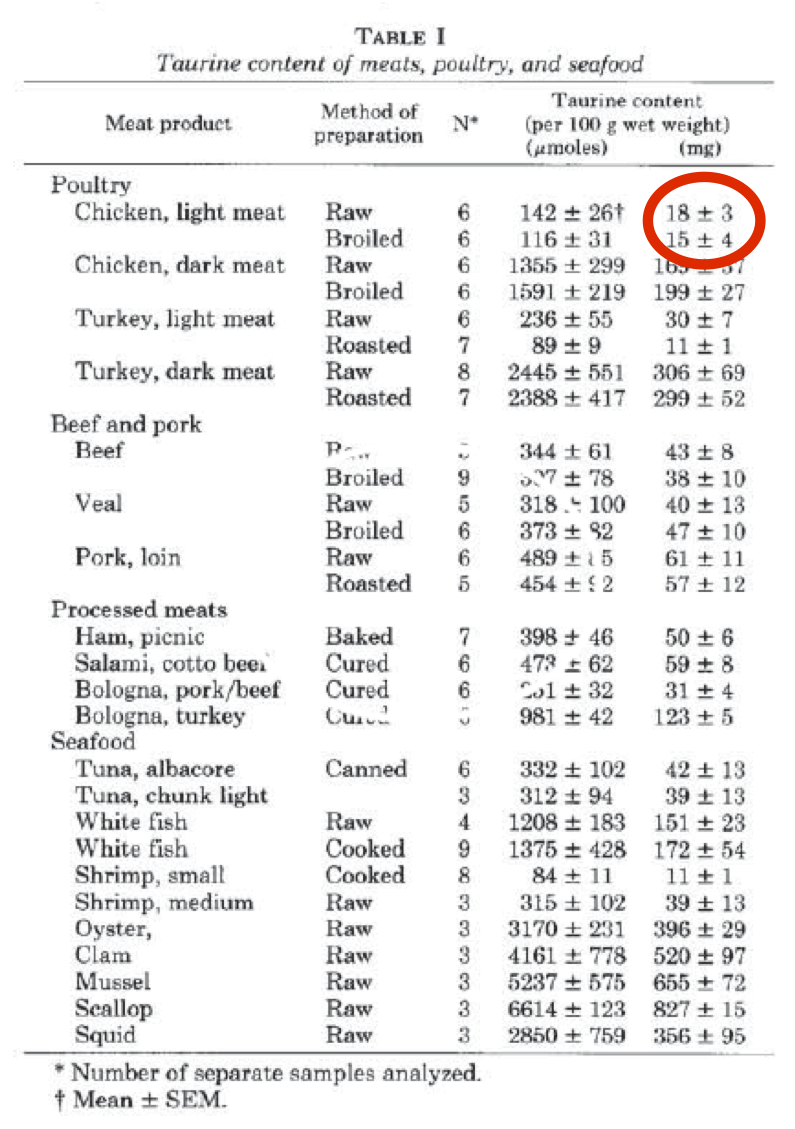
Poultry breast meat is special when compared to poultry leg/wing meat. It has the lowest taurine content of any meat. If you feed your cat on chicken fillets she will develop dilated cardiomyopathy. If the cardiomyopathy allows the generation of an atrial thrombus which breaks off the syndrome produced (iliac thrombosis) is terminal, whatever hope your vet might cautiously suggest re "treatment".
On the plus side DCM from taurine deficiency is completely reversible. Mini was re-scanned recently by the same cardiologist and now has a normal heart. She has been off of frusemide for months.
Taurine comes by the kilo as a sports supplement. I put a pinch of it on her food each night.
Due to my initial thoughts before getting the diagnosis I'd changed her on to beef mince yet again and was going to put up with paddling in cat vomit on the kitchen floor occasionally. Didn't happen. No more vomiting on beef. Huh?
So now all of our cats are on beef mince.
Moral of the story:
Don't feed you cat on chicken fillets.
That was a very, very near miss and (another) lesson to me that I do not have all of the answers! Occasionally reality bites you.
Peter
Addendum. There is a similar known problem with feeding whole ground rabbit carcasses to cats (of which I was aware, but had missed the selective taurine deficiency in white poultry meat):
Rabbit Carcasses for Use in Feline Diets: Amino Acid Concentrations in Fresh and Frozen Carcasses With and Without Gastrointestinal Tracts
Addendum. There is a similar known problem with feeding whole ground rabbit carcasses to cats (of which I was aware, but had missed the selective taurine deficiency in white poultry meat):
Rabbit Carcasses for Use in Feline Diets: Amino Acid Concentrations in Fresh and Frozen Carcasses With and Without Gastrointestinal Tracts