Deuterium-reinforced polyunsaturated fatty acids protect agains atherosclerosis by lowering lipid peroxidation and hypercholesterolemia
The APOE*3-Leiden mice were reared on non specific chow. At 12 weeks of age (Time -4) they were put on to something derived from the AIN-93M diet. All lipids were all supplied as methyl esters of fatty acids, not triglycerides. It contained 1.2% of calories as LA and 9% of calories as sucrose. The intervention group had exactly half of the 1.2% of LA calories supplied in the form of deuterium stabilised, ROS peroxidation resistant D2-linoleic acid.

First aside: Mouse model. This current model, the APOE*3-Leiden mouse, is a model. It's not as totally useless as an APOE total knockout or an LDL receptor knockout model but it's still nothing like a real mouse or like a real human. Mouse lipoprotein management is not like human lipoprotein management. They do not have a cholesterol ester transfer protein. The Leiden mouse has this human gene engineered in. It also has a selective APOE*3 knockout to give a mild elevation of LDL. The end result is a model which has numbers on a lipid panel which look a bit more like a human with metabolic syndrome than the average extreme knockout mouse model.
But it's not a human with metabolic syndrome. It's an APOE*3-Leiden mouse and if you found a cure for its "atherosclerosis" you would have a great tool for helping APOE*3-Leiden mice. Would it translate to helping humans with metabolic syndrome? Hahahahahahahahah bonk. End aside.
Second aside: Does LDL cause atherosclerosis? Hahahahahahahah, bonk. To anyone with any sense atherosclerosis is a response to injury where IGF-1 delivered by platelets attaching to the injured endothelium causes media hypertrophy to reinforce the site of injury. This is accelerated if systemic hyperinsulinaemia also acts as an agonist on those IGF-1 receptors. It can almost certainly be enhanced by delivering lipid peroxides such as 4-HNE, 13-HODE and 9-HODE, though their effects are very, very complex. I'm perfectly willing to believe that any genetic engineered tweak in to a mouse which increases the persistence of linoleic acid containing lipoproteins in the plasma allows time for that LA to spontaneously oxidise and accelerate what looks a bit like atherosclerosis, in the model. Just my view. End second aside.
OK. On to the paper:
The APOE*3-Leiden mice were reared on non specific chow. At 12 weeks of age (Time -4) they were put on to something derived from the AIN-93M diet. All lipids were all supplied as methyl esters of fatty acids, not triglycerides. It contained 1.2% of calories as LA and 9% of calories as sucrose. The intervention group had exactly half of the 1.2% of LA calories supplied in the form of deuterium stabilised, ROS peroxidation resistant D2-linoleic acid.
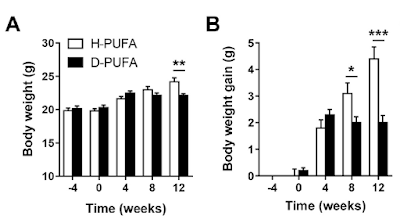
They were fed these diets for four weeks. At that point (Time zero) 0.15% by weight of reagent grade cholesterol was added to both diets (otherwise the model doesn't work to get the essential-for-funding lipid lesions, no sniggering at the back. It's a model). This "western" diet, which only differed from the run-in diet by the added 0.15% reagent grade cholesterol, caused/allowed some weight gain over the following 12 weeks but less in the D2-LA supplemented group than the normal LA group:

No weight gain on either of the run-in diets, followed by lots of gain in the "normal" LA diet but not the D2-LA diet once the cholesterol was added.
Why?
Why should adding 0.15% of cholesterol produce such diverging weight gains?
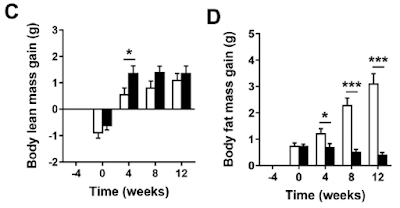
Even more exciting is if you look at lean mass vs adipose mass:

Adding just 0.15% cholesterol produced a marked fat mass gain in the normal LA mice and a trend downward in fat mass for the ROS protected D2-LA mice.
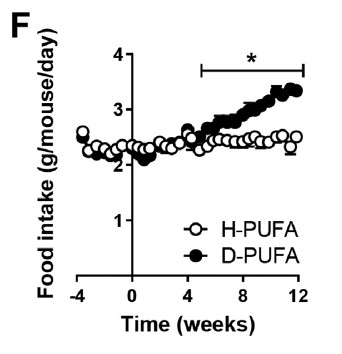
On top of that the D2-LA mice started eating extra during the period of fat loss. A lot extra:

Soooooooo. What is going on?
Well, the first thing to realise is that during the four week run-in period after the replacement of chow by the AIN-93M derived diets there were already changes which didn't show in total body weights (graphs A and B). The mice, with or without deuterium stabilised LA, all lost muscle mass and all gained fat mass during those weeks, just under a gram of each. So, even without the reagent grade cholesterol, changes were already on going from a "normal" mouse phenotype on chow towards a "skinny-fat" phenotype on an AIN-93M-like diet. That's clear in graphs C and D. Possibly from the sucrose but there's no way of telling that from the paper.
The changes were on-going before the diets were "westernised" by the addition of 0.15% of reagent grade cholesterol. I suspect that the addition of the cholesterol is a red herring.
Let's go on to look at food intakes.
The mice on the deuterated LA eventually began eating more than those on the standard LA. This became statistically significant at about week five.

Any mouse which is eating extra and losing adipose tissue is either showing malabsorption or uncoupling.
I'll buy the uncoupling, but then I would.
Why the delay to the onset of starting to eat extra? Is there a delay in uncoupling onset? Not necessarily. A normal mouse uses a significant percentage of its caloric intake to generate heat in its brown adipose tissue. There is no need to increase food intake while ever the purported uncoupling from deuterated D2-LA is generating less heat than is needed to maintain body temperature. As heat production increases over time it begins to exceed this essential minimum and so comes to represent a "calories-out" in excess of what is merely needed to keep warm. At this point an increase in food intake becomes necessary to balance the heat lost by supra-physiological uncoupling.
If this is correct, and that's a big if, there is clearly an on-going progressive increase in uncoupling with time. The logical explanation is that there is a progressive increase of deuterated D2-LA in tissues and/or being used for beta oxidation.
How might deuterated, ROS resistant LA, facilitate uncoupling?
I don't know, so it's time for some routine wild speculation. If we could answer that one question the whole scenario becomes straight forward. Sadly it is not at all obvious why D2-LA should facilitate uncoupling. Here's my current best shot. If I think of something more plausible I'll post again:
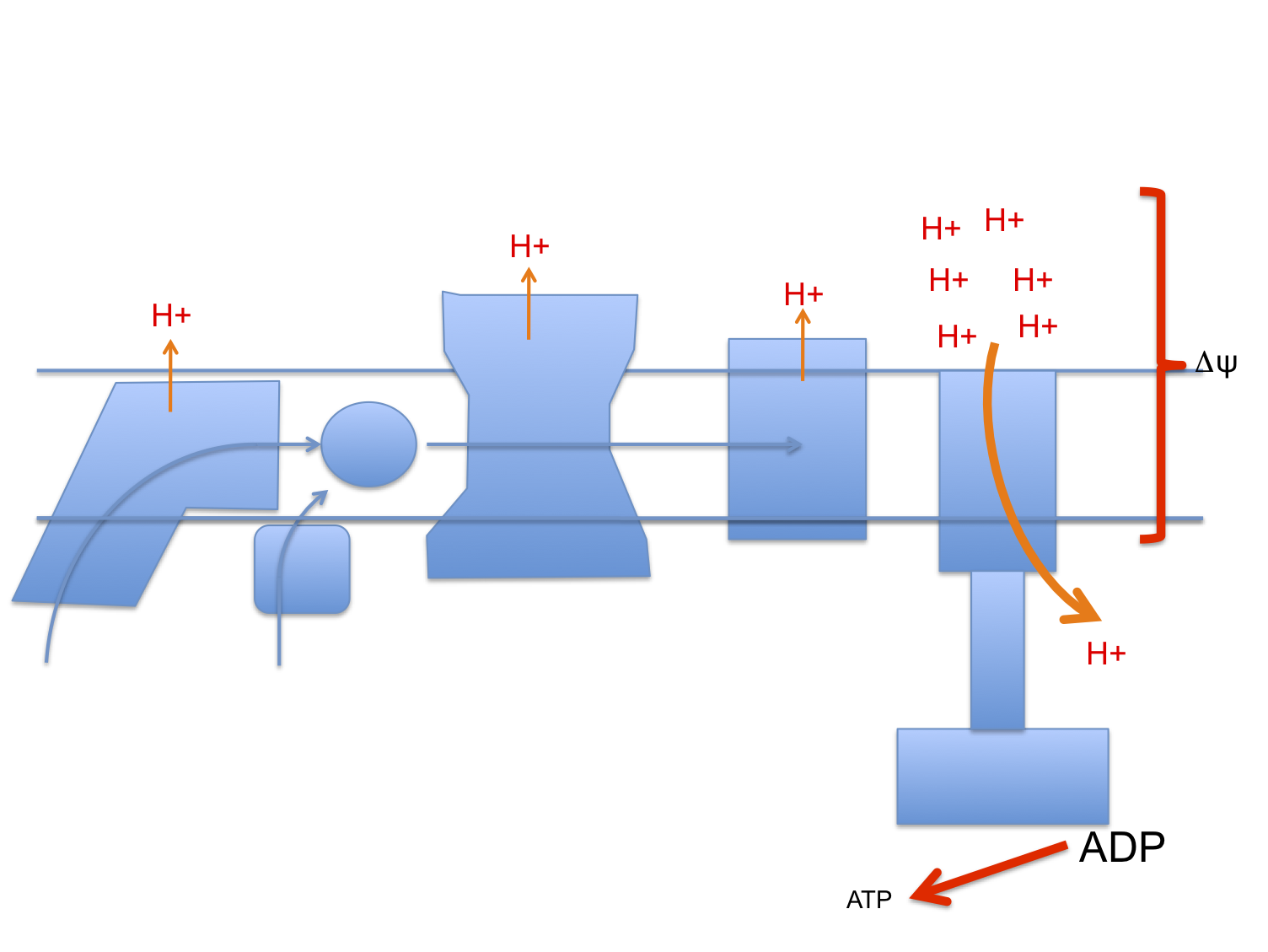
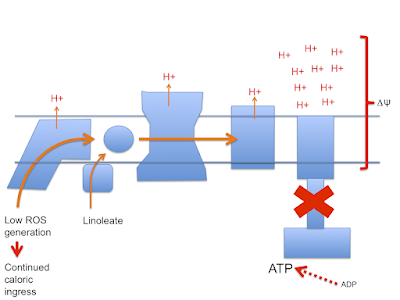
Let's assume that linoleic acid, whether deuterated or native, allows excess calories in to a cell. This is the doodle from a few posts ago:

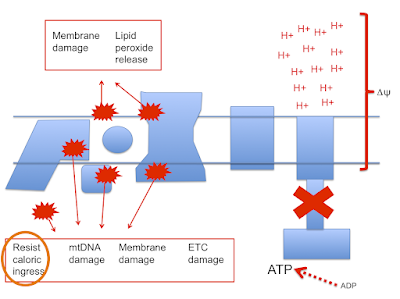
which then leads to this doodle:

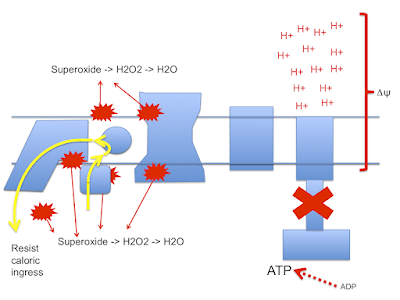
and this doodle:

This begs the question as to how much damage (signalling?) is done by the stray electrons, how much by superoxide, by hydrogen peroxide or how much is actually mediated by the lipid peroxides generated from linoleic acid per se. Which is the most important mediator?
Let me suggest that D2-LA allows the excessive delta psi, which facilitates both reverse electron transport and pathological ROS generation. As in the previous post the high delta psi eventually allows D2-LA to drive RET at the cost of, via high delta psi, allowing electrons to be lost from the ETC to oxygen at abnormal sites, forming superoxide. Under D2-LA this superoxide has very limited ability to contact oxidisable native linoleic acid.
So now we look more like this with ;

There is a surfeit of ATP, high delta psi and availability of either LA and/or D2-LA to facilitate uncoupling combined with minimal damage to the ETC. You do have to have a source of 4-HNE or a related "damage marker" to facilitate uncoupling but it doesn't need much.
I can't see any more straightforward technique for D2-LA to uncouple. If there is one and it's clear how it works, that would be great. Currently this is the best I can do.
Summary: D2-LA allows uncoupling. That explains everything, but the mechanism for the uncoupling is obscure and I'm guessing.
NB I was also trying to explain to myself why the control group got fat. I don't think they did. A total weight gain of 5g over 12 weeks to give a final weight of 25g sounds like a normal mouse to me. It's the slim mice eating extra food to maintain that slim bodyweight that are abnormal.
Ultimately the paper poses the question: What determines whether a cell deals with excess calories by sequestration to storage vs uncoupling. Obviously this is insulin signalling. But is it an oxidation product of linoleic acid which controls insulin signalling? Are we simply looking at a situation of absolutely suppressed insulin signalling, due to D2-LA being "too" stable?
Peter
Final addendum/aside. There is a claim on 'tinternet, un referenced, that low dose oral deuterium oxide in mammals causes weight loss, rather than the death which is the normal result of high dose exposure. Could D2O trigger uncoupling irrespective of LA type and the catabolism of D2-LA be a simple source of deuterated water by oxidation of the D2-LA? I doubt this but the idea is still a potential explanation. I have hunted support/refutation for this without success.