Redox-induced activation of the proton pump in the respiratory complex I
Here is Figure 1, it's the inset we're interested in:

and here is the inset. I've taken the liberty of inserting arginine R216 as shown in Figure 6 (left panel) and as mentioned in the text section "Electrostatic coupling elements". Which is what we want to know about.

To make sense of this it's easiest to break it down in to three sections, each representing a specific process. We can start with the aspartate D139 which is protonated and hydrogen bonded to histidine H38, like this. I've faded the rest out:
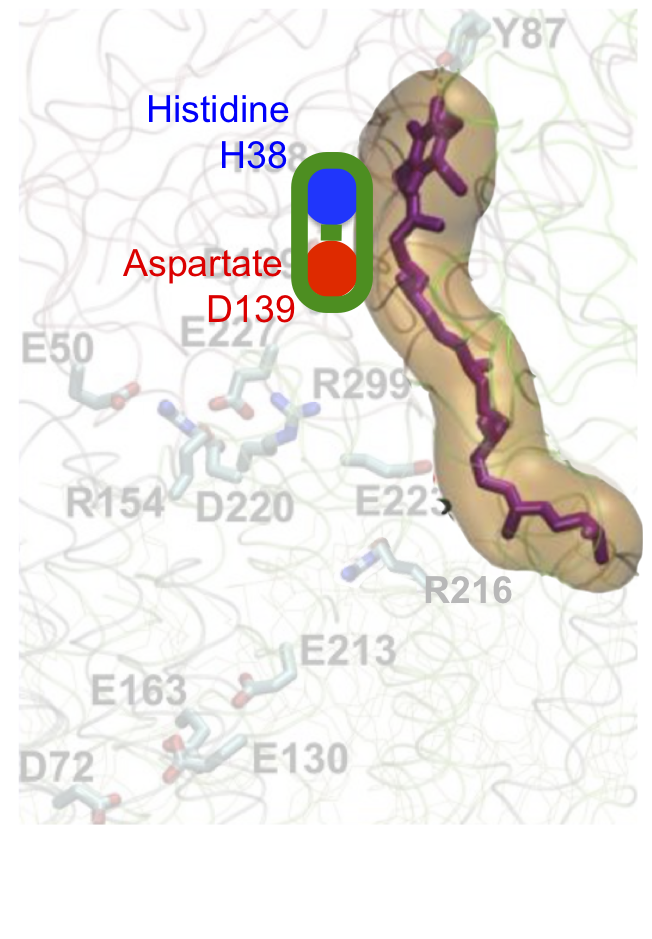
Two electrons are delivered to CoQ from NADH and nothing happens. A few picoseconds later one electron on CoQ "steals" a proton from histidine H38 (along with a second proton, for the second electron, taken from the Tyrosine Y87 just visible at the top of the image. I've left this out for clarity) to form reduced CoQ2H:

Histidine H38 immediately replaces its lost proton by "stealing" it from aspartate D139. This aspartate becomes negatively charged and alters the protein conformation to move itself downwards (in the diagram)
taking an area of negative charge with it:

Now we can move on to step two and add in some more important amino acids. These red circles are all glutamates and the blue circles are all arginines:

The combination of change to surrounding protein shape with the localisation of the negative charge on aspartate D213 forces the combination of the arginines with the glutamates in to electrostatically bonded pairs shown as green ovals. The dotted green oval is my guess, the two solid ones are specified in the paper:

which repositions the polar amino acids like this:

Quite how this rearrangement forms a proton channel is unclear (or whether protons are simply transferred from amino acid to amino acid without a water channel forming, there doesn't appear to be a water channel modelable, yet) but the paper suggest it does so and the negative charge zone encourages protons to transfer from the bulk solvent of the cytoplasm to the centre of the complex:

The final step involves these amino acids, mostly glutamates with an aspartate D72 at the end of the chain:

The conformation change in the protein structure moves these amino acids towards the source of protons

and puts them in to a microenvironment which makes them highly avid to gain a proton, which they do:

Working on the basis of electrostatic coupling between well investigated antiporter-like pumping modules it looks to me very much like the protonation of aspartate D72 provides the "kick" to the messy fourth proton pathway between NuoN and the small membrane subunits NuoA, J and K:

Up until now pretty much all of what I have described is what is reported in the paper from their extensive modelling work. Now I'm going to speculate.
I don't think these NuoH protons go anywhere towards being transported to the periplasm. I think they go back the way they came. One of the crucial steps after the reduction of CoQ to CoQ2H is the restoration of the protonation of the amino acids which have provided the protons to join with the electrons on CoQ to give a neutral molecule. It's not at all clear where these replacement protons might come from, so I feel free to speculate. In this particular complex I example we are talking about reprotonating aspartate D139 and tyrosine Y87, either side of the CoQ binding pocket. Like this:

In particular the restoration of protonation of aspartate D139 will allow it to return to a hydrogen bonded to histidine H38 position and allow protein conformation to return to the baseline level overall, leaving the system ready to fire again.
This speculation is compatible with a non proton pumping function of the half-channel in NuoH/Nquo8 but a crucial function in transmitting the energy from CoQ reduction to the antiporter modules. It also gives a speculative mechanism for the reprotonation of the amino acids deprotonated in CoQ reduction. I like the idea. It makes sense (which clearly does not mean it is correct!).
I would also guess that in an optimised system that only two protons are used to effect the aspartate D72 "kick" and these two protons are the ones which are returned to neutralise the changes around the CoQ binding pocket.
I'm now set to try and work out what evolution was doing to set up a pre-adaptation to this rather bizarre system. Fingers crossed.
Peter












.gif)






















