The hypothesis paper
New Control of Mitochondrial Membrane Potential and ROS Formation – A Hypothesis
mentions some factors which might flood mitochondria with Ca2+. Vasopressin gets a mention, again an excellent pressor drug for intractable hypotension, if you don't mind the calcium.
But of course the hormone we all want to ask about is insulin. So you go to Pubmed and search on "insulin calcium ROS"
which brings up this as pretty much the first hit
I haven't read the paper, all I wanted to know was whether insulin behaved as a "stress" hormone as regards Ca2+:
"The insulin-dependent Ca(2+) released from IP3R of skeletal muscle also promotes mitochondrial Ca(2+) uptake."
"The insulin-dependent Ca(2+) released from IP3R of skeletal muscle also promotes mitochondrial Ca(2+) uptake."
It does. Okay, slightly more interesting (and more thoroughly read) is this one:
Insulin enhances mitochondrial inner membrane potential and increases ATP levels through phosphoinositide 3-kinase in adult sensory neurons
A lot of cautions yet again. It's neural tissue, which is not a typical insulin sensitive tissue and the paper is old enough that measuring mitochondrial membrane potential was a bit more difficult than ordering a fluorescent dye kit form some generic laboratory supply company.
However it does seem that in something resembling real cells that insulin not only increases delta psi but it also increases ATP levels. Of course we don't know whether the extra ATP comes from glycolysis or increased ox phos from this paper. We do know that it increases.

The insulin concentration used here is 1.0nM which is essentially equivalent to maximal physiological concentration in the aftermath of a meal of modern junk food. Here is the pattern of mitochondrial hyper polarisation at exposure to increasing insulin concentrations. Using 0.75nM is absolutely physiological (if you eat junk food). The glucose used appears to be in the region of 10mM (Ham's F12 medium) and the medium is serum (ie fatty acid) free:

Certain things are clear. There is no dose-response to insulin. Even physiological levels produce a maximal rise in membrane potential. If the pre-insulin membrane polarisation is around 100mV (the technique to assess polarisation couldn't give an absolute value in 2004) then insulin will double this. That seems pretty well certain to generate a membrane polarisation well over the 140mV which will generate copious ROS.
Next thing from here (again)
is that insulin signaling, as assessed by the proportion of Akt which is phosphorylated, is also maximal at exposure to insulin at modestly greater than peak physiological levels (here insulin at 5nM with glucose at ~5mM, some bovine serum and glutamate):

This leaves me with an unanswered question.
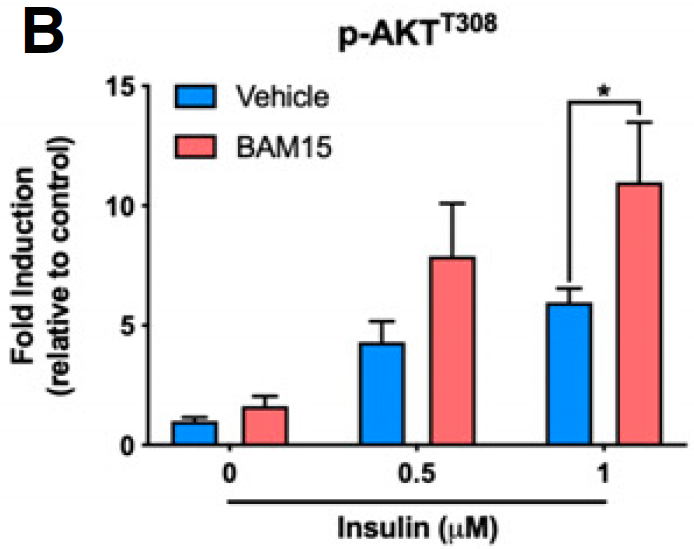
We know that we can increase Akt phosphorylation during massively supra-physiological insulin exposure by simply limiting delta psi with agents such as BAM15 or DNP. This is because we limit the ROS generation which is needed to induce insulin-induced insulin resistance, allowing a little extra pAkt to be formed before ROS exceed a critical threshold. We also know that DNP at in-vivo concentrations does the opposite, it reduces insulin signaling. I won't re-cite the same old papers.
What I would like to know is what the oxidation of linoleic acid does to pAkt levels under physiological insulin exposure, compared to palmitic acid. If it is the generation of ROS which limits the rise in pAkt then the lower ROS generation under linoleic acid oxidation should allow more pAkt formation, with enhanced insulin signalling under physiological conditions, which is essential for the development of obesity.
One of my core tenets is that LA limits the normal resistance to insulin signaling mediated by ROS generation. My opinion is the LA causes insulin resistance only once insulin signaling augmentation has produced distended adipocytes which release FFAs in the face of elevated insulin/glucose. In the pre-obese state LA facilitates insulin signaling. Otherwise you wouldn't get fat.
To my knowledge no one has looked at Akt phosphorylation when specific fatty acids are being metabolised under reasonably physiological insulin and glucose concentrations. It would be great to know if LA allowed more pAkt to be formed.
I'd guess that this would be the case.
I have a few more speculations about FFAs, glucose/insulin and ROS which I might leave for another post as this one is getting unwieldy, yet again.
Peter