Insulin -> Insulin Receptor (IR) -> G coupled protein -> NADPH oxidase 4 (NOX4) -> ROS
These ROS are the spark which triggers insulin signalling.
They act to inhibit assorted protein tyrosine phosphatases (PTPs). With PTPs disabled the normally suppressed auto-phosphorylation of the IR activates and its self activation allows calories to flood in to the cell. So NOX4 starts the process. It's a eukaryote system, there is nothing fundamental about it, just as insulin signalling is an eukaryote system. But it uses ROS because they are fundamental.
Once glucose calories are entering the cell they are supplied to the mitochondria which generate ROS by the rise in F:N ratio, triggered by the glycerophosphate shuttle (my opinion) converting glucose derived cytoplasmic NADH into mitochondrially targeted FADH2. My expectation is that this degree of ROS generation will keep the PTPs suppressed and so the insulin receptor phosphorylated/active while ever the cell is using glucose and so still wants glucose calories to enter.
Once glucose calories are entering the cell they are supplied to the mitochondria which generate ROS by the rise in F:N ratio, triggered by the glycerophosphate shuttle (my opinion) converting glucose derived cytoplasmic NADH into mitochondrially targeted FADH2. My expectation is that this degree of ROS generation will keep the PTPs suppressed and so the insulin receptor phosphorylated/active while ever the cell is using glucose and so still wants glucose calories to enter.
These ROS are physiological and represent the normal control and maintenance of insulin signalling at peak levels. The next step is the use of ROS to disable insulin signalling.
There are times when it is necessary to actively suppress insulin signalling. This is most easily visualised by considering a cell which has received more than enough insulin mediated calories to meet its needs. Under these circumstances there is a surfeit of ATP which activates a negative feedback acting on ATP synthase. The inhibition of ATP synthase means that delta psi is no longer being dissipated so it will increase. At values above 140mV the rate of ROS generation increases exponentially and reaches levels that will directly act on the proteins of the insulin receptor and signalling cascade to de-activate them.
This is beautifully illustrated in this paper from 1974, previously discussed here
It's tracking the evolution of CO2 from radiolabelled glucose and using H2O2 as a direct replacement for insulin to facilitate glucose uptake/oxidation by adipocytes.

It is very clear that 0.01mM of H2O2 initiates glucose uptake/oxidation, ie with absolutely no insulin present then 0.01 mM of H2O2 has the signalling action of a low dose of insulin. I think of this as equivalent to the NOX4 action. There is a level of H2O2 at around 0.3mM which performs the function of peak insulin exposure. When H2O2 exposure is further increased to 4mM or 5mM then the H2O2 disables its own ability to replace the function of insulin.
Let's make this absolutely clear, insulin is in no way fundamental. It can be completely replaced by H2O2 which, in common with superoxide, is the core signalling system and is ubiquitous as a growth/reproductive signal going right back to bacteria, preserved in their behaviour today. H2O2 is not an insulin mimetic, it is the core signal. Suppressing this ROS signal in modern bacteria, which never use insulin, inhibits their growth and reproduction.
Aside: No one has yet worked out how bacteria generate superoxide or H2O2 today. It doesn't seem to be via NOX enzymes which are, sadly, purely eukaryotic. I say sadly because they are extremely simple enzymes with a six helix tube through the cell membrane containing two Fe moieties as a "wire" to carry electrons to extracellular O2 from a bolted-on NADPH oxidase within the cell. If we ignore the multiple control systems which can also be added then it's very simple. The core of NOX just looks primordial. But it isn't. Sigh. End aside.
I hope I have established that there is a primordial signalling system which uses low concentrations of ROS to initiate nutrient uptake and usage to a certain maximal effect, above which a negative feedback using the same ROS disables further nutrient uptake/utilisation.
I've left fatty acid oxidation out of this narrative to keep it simple.
All of which leads us back to 4-HNE.
We can accept that the NOX4 minor contribution and the mitochondrial major contribution to ROS are generated in absolute proximity to the lipid membranes. All lipid membranes have functional needs for PUFA for structural purposes and if these are directly abutted to the electron transport chain complexes which are producing the above ROS signals then we can expect a proportion of the ROS to interact with those PUFA. The best studied end product is 4-HNE.
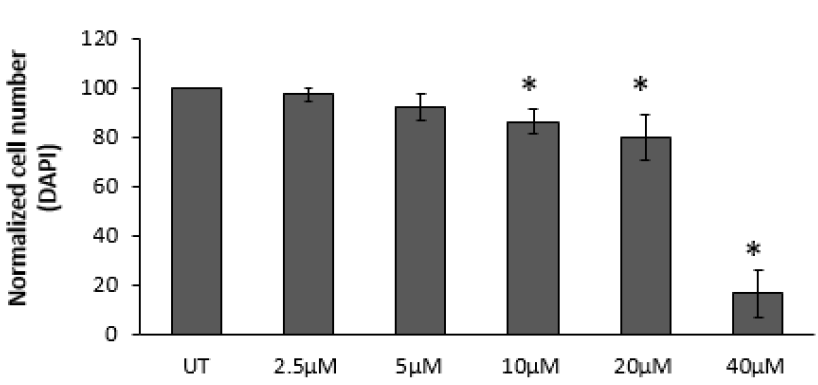
Superoxide is a poorly mobile signalling molecule ideal for short distance signalling but with limited ability to cross lipid membranes. H2O2 is more stable and better able to cross lipid membranes so makes a good intermediate distance signal within a cell. 4-HNE carries the same information about the state of the ROS generation from the electron transport chain (pax NOX4) but is stable enough to be transported through the blood stream in measurable and modifiable concentrations. Because it carries the same information as superoxide/H2O2 it should come as no surprise it elicits the same response. In fact I view it as an amplifier of the ROS signal. It seems that 0.1μM 4-HNE can give a similar effect on adipocyte lipid accumulation as 0.3mM (ie 300μM) H2O2. Roughly. And anything over 5μM 4-HNE disables insulin signalling, equivalent to 5mM H2O2.
Obviously Protons makes this simple story a little more complex as you add in fatty acids and the influence of linoleic acid which both reduces and increases ROS generation as well as being the core substrate for generating 4-HNE per se, but I think this will do for today.
The above description makes the whole system look static. It's not. All of it oscillates, delta psi and ROS generation. You can see why.
Peter
Throw away thoughts:
If ROS are a growth signal, so too should be 4-HNE. Anyone can Pubmed "cancer + 4-HNE".
Also H2O2 is a biological warfare molecule. Macrophages "throw" H2O2 at bacteria to kill them. Any PUFA in the area of the battleground will generate 4-HNE. This is a medium distance signal to recruit more macrophages to join the fight. These are not directly related to metabolism but clearly important. Actually, you could ask whether high levels of both H2O2 and 4-HNE kill bacteria metabolically. There's a thought.
Then you can get in to plasma 4-HNE and it's effects on lipoprotein PUFA components. 4-HNE is a direct generator of ROS in its own right (it's function is as an amplifier of the ROS signal after all) so filling lipoproteins with linoleic acid while elevating plasma 4-HNE will attract macrophages to the extra 4-HNE wherever those lipoproteins stick. Like arterial/arteriolar walls. Stroke? Heart attack?
Hmmm. All fun stuff. Blame your cardiologist.