I've been sent a link to this study:
Body fat loss and compensatory mechanisms in response to different doses of aerobic exercise—a randomized controlled trial in overweight sedentary males
Bottom line: Moderate exercise produces equal weight loss to greater exercise. I'll come back to this later.
I commented in the last post that I consider leaving food on your plate is a very reasonable surrogate for experiencing reduced hunger. This shows up as weight loss. When we look at the converse, long term "accidental" weight gain, my perspective is the same. Eating an extra portion is a surrogate for responding to hunger. Long term hunger = Long term weight gain.
Let's say that again:
Fat people are fat because they are affected by hunger. Not gluttons. Not lazy. Hungry.
If people are going to lose weight on a long term basis they can only do this if they are not hungry. If they are not hungry they will reject food, establish an energy deficit and lose weight.
I may believe in CICO, but that CICO is controlled by hunger.
Back to the study. As the authors, struggling for a moment of lucidity, say:
"However, on the basis of the present findings, we propose that the introduction of a moderate dose of exercise may actually lead to an increase in NEAT without any increase in EI resulting in a “bonus effect,” whereas a higher dose of exercise may lead to an increase in EI and, thereby, a degree of compensation and less than expected loss of FM"
As clear as mud. Let's translate and simplify:
“… a moderate dose of exercise may actually lead to an increase in CALORIES OUT without any increase in HUNGER ... whereas a higher dose of exercise may lead to an increase in HUNGER [ie more eating] and … less than expected loss of fat mass”.
Exercise makes you hungry, certainly if you over-do it.
This is the authors' conclusion. I think they are correct. Other explanations are possible but they if miss the hunger component they are not of a great deal of help to anyone who wishes to understand obesity, even if they happen to be factually correct.
Peter
Monday, August 31, 2015
Sunday, August 30, 2015
Confirmation Bias in my head
Sometimes you get a reminder that you suffer from Confirmation Bias.
I do.
This morning I went back and dug out the study from Aberdeen:
Which carried the phrases:
"…participants were offered a fixed energy intake of 2000 kcal/d"
"In the isocaloric study, despite being served food of the same energy content, intake was slightly lower (66 kcal/d) and weight loss greater (7.2 ± 2.3 vs. 4.7 ± 1.0 kg in 4 wk, P less than 0.05), on the HF-LC diet after correction for unconsumed food"
"In the isocaloric study, despite being served food of the same energy content, intake was slightly lower (66 kcal/d) and weight loss greater (7.2 ± 2.3 vs. 4.7 ± 1.0 kg in 4 wk, P less than 0.05), on the HF-LC diet after correction for unconsumed food"
Feed overweight men 2000kcal/d of a mixed diet and they eat it all. Feed the same men 2000kcal/d of a mildly ketogenic diet (roughly ++ on ketostix) and they refuse to eat all of the food. What they reject gives a weight loss surfeit compared to when they were on the mixed diet.
There's no evidence they went to the gym on the quiet but the study protocol probably asked them not to do this. Assuming they have gyms in Aberdeen.
I don't give a monkey's about appetite score by VAS, lack of metabolic advantage in a study using 140g/d of carbs etc. Food left on plate = Not hungry.
How much simpler can it be?
This was triggered by an article linked to by Rose Nunez Smith via FaceAche which piqued my interest and which I read right through to the end. The article kept saying things which made sense and fitted with my view of reality. There was no author on the end so I went up to the top only to find it was Gary Taubes.
Well, it made me laugh.
It's good there are people writing for the NYT Sunday Review who point out what really matters. Bugger any metabolic ward study.
BTW re Aberdeen, with ketones at ++ it would have been interesting to know what the FFA levels were like but the arithmetic cited in the study suggests we didn't have a lot of UCP activity during this few weeks of ++ ketostix eating.
Peter
Tuesday, August 25, 2015
Starchy stable isotopes? I don't think so!
Have a read at this statement from Hardy et al 2015:
“…stable isotope analyses indicate a mainly carnivorous diet for Neanderthals; a wider range of isotopic values have been observed in contemporary Middle Pleistocene H. sapiens (Richards and Trinkaus 2009), indicating that considerable differences in the levels of starch consumption existed between these two species.”
Now, if you read this I think you might be led to believe that stable isotope analysis indicates that Neanderthals were carnivores and H sapiens ate a different amount of starch to a carnivore. I feel the implication of this sentence is that H sapiens ate "more-than-zero starch" during the Middle Pleistocene.
You would believe wrongly. Did you check the reference? No? Naughty. Richards and Trinkaus (2009) actually say this:
“As the method only measures protein intake, many low-protein foods that may have been important to the diet (i.e., high caloric foods like honey, underground storage organs, and essential mineral and vitamin rich plant foods) are simply invisible to this method.”
The data do not deny starchivory. But the data equally do not in any way support its occurrence. Starch, fruit and honey are invisible on stable isotope analysis. This is a gross mis-citation of Richards and Trinkaus by Hardy et al. Never believe stuff like this without checking the refs. Easy when it is a freebie in PLOS. What do Richards and Trinkaus actually say about diets of carnivorous Neanderthals vs H sapiens? Try this:
“There are now enough isotopic data to see patterns in the data, and they show that the Neanderthals and early modern humans had similar dietary adaptations, obtaining most of their dietary protein from animals, although some of the early modern humans obtained significant amounts of their protein from aquatic, and not just terrestrial, sources.”
You can tell H sapiens ate fish because aquatic food chains are long. The longer the food chain the greater the effect visible in stable nitrogen isotopes. They make fish eating carnivores look like hyper-carnivores. That's how they show up in the paper. Had humans eaten any significant amount of protein rich plants (hazel nuts get cited as a possibility) it would show a lower stable nitrogen ratio. There is no evidence for this.
Did early humans consume starch to grow their brain size? Stop laughing! No one knows, certainly to the point where a starchivorous paper has to mis-cite a completely non-supportive paper as being actually supportive of their rubbish hypothesis.
I love it.
Did you hear the one about Jennie Brand-Miller? Passthecream linked to this gem in the comments of the last post. Some things are just too funny not to share. Have a giggle. J B-M is second author on the starch-is-needed-to-grow-brains paper...
Peter
“…stable isotope analyses indicate a mainly carnivorous diet for Neanderthals; a wider range of isotopic values have been observed in contemporary Middle Pleistocene H. sapiens (Richards and Trinkaus 2009), indicating that considerable differences in the levels of starch consumption existed between these two species.”
Now, if you read this I think you might be led to believe that stable isotope analysis indicates that Neanderthals were carnivores and H sapiens ate a different amount of starch to a carnivore. I feel the implication of this sentence is that H sapiens ate "more-than-zero starch" during the Middle Pleistocene.
You would believe wrongly. Did you check the reference? No? Naughty. Richards and Trinkaus (2009) actually say this:
“As the method only measures protein intake, many low-protein foods that may have been important to the diet (i.e., high caloric foods like honey, underground storage organs, and essential mineral and vitamin rich plant foods) are simply invisible to this method.”
The data do not deny starchivory. But the data equally do not in any way support its occurrence. Starch, fruit and honey are invisible on stable isotope analysis. This is a gross mis-citation of Richards and Trinkaus by Hardy et al. Never believe stuff like this without checking the refs. Easy when it is a freebie in PLOS. What do Richards and Trinkaus actually say about diets of carnivorous Neanderthals vs H sapiens? Try this:
“There are now enough isotopic data to see patterns in the data, and they show that the Neanderthals and early modern humans had similar dietary adaptations, obtaining most of their dietary protein from animals, although some of the early modern humans obtained significant amounts of their protein from aquatic, and not just terrestrial, sources.”
You can tell H sapiens ate fish because aquatic food chains are long. The longer the food chain the greater the effect visible in stable nitrogen isotopes. They make fish eating carnivores look like hyper-carnivores. That's how they show up in the paper. Had humans eaten any significant amount of protein rich plants (hazel nuts get cited as a possibility) it would show a lower stable nitrogen ratio. There is no evidence for this.
Did early humans consume starch to grow their brain size? Stop laughing! No one knows, certainly to the point where a starchivorous paper has to mis-cite a completely non-supportive paper as being actually supportive of their rubbish hypothesis.
I love it.
Did you hear the one about Jennie Brand-Miller? Passthecream linked to this gem in the comments of the last post. Some things are just too funny not to share. Have a giggle. J B-M is second author on the starch-is-needed-to-grow-brains paper...
Peter
Monday, August 17, 2015
Sweden's dietitian advice? No thank you.
Hot off the press from Uppsala and Stockholm:
A high energy intake from dietary fat among middle-aged and older adults is associated with increased risk of malnutrition 10 years later.
"Contrary to what was expected, a high energy intake from total fat, saturated fat and monounsaturated fat among middle-aged and older adults increased the risk of exhibiting malnutrition 10 years later. However, this applied only to individuals with a BMI < 25 kg/m2 at the baseline. In conclusion, these findings suggest that preventive actions to counteract malnutrition in older adults should focus on limiting the intake of total fat in the diet by reducing consumption of food with a high content of saturated and monounsaturated fat."
Repeat after me. Association does not prove causation. How anyone dare suggest an experimental intervention on a large subpopulation of their nation based on an observational association within a subgroup of the target population is beyond me. How dare they?
It must be embarrassing to be a dietitian in Sweden nowadays but this sort of intervention recommendation is not going to decrease the stupidity index of mainstream dietary advice.
Hopefully sensible people will continue to ignore them!
Peter
A high energy intake from dietary fat among middle-aged and older adults is associated with increased risk of malnutrition 10 years later.
"Contrary to what was expected, a high energy intake from total fat, saturated fat and monounsaturated fat among middle-aged and older adults increased the risk of exhibiting malnutrition 10 years later. However, this applied only to individuals with a BMI < 25 kg/m2 at the baseline. In conclusion, these findings suggest that preventive actions to counteract malnutrition in older adults should focus on limiting the intake of total fat in the diet by reducing consumption of food with a high content of saturated and monounsaturated fat."
Repeat after me. Association does not prove causation. How anyone dare suggest an experimental intervention on a large subpopulation of their nation based on an observational association within a subgroup of the target population is beyond me. How dare they?
It must be embarrassing to be a dietitian in Sweden nowadays but this sort of intervention recommendation is not going to decrease the stupidity index of mainstream dietary advice.
Hopefully sensible people will continue to ignore them!
Peter
Methyltransferase in methanogens
The archaea developed rather differently from the Ech driven bacteria. I want to look at them from the energetic point of view so it seems reasonable to start with this image from the first of the Life series posts, back in February:

This has CH3-SH (used at two points in the process) driving acetate formation for cell carbon generation. We know this works because Huber and Wächtershäuser demonstrated the abiotic generation of activated acetate from CO and CH3-SH in the presence of an FeS/NiS slurry. No enzymes, no cofactors, no structure. Energetically, it works. I now want to speculate wildly about other uses of CH3-SH in the development of methanogens and the evolution of methyltranseferase, the archaeal alternative to Ech. Let's get rid of the carbon fixation doodles. The location of CH3-SH might need to change as ideas develop:

In the background to the following speculation we still have a FeNi hydrogenase "visiting" a proton channeling pore to generate reduced ferredoxin using a localised low pH region just inside the cell. In the archaea I'm assuming there is no preformed Na+ pump because the protein translocating precursor never jammed up so never pumped Na+ ions. There was no drop in intracellular Na+ and the FeNi hydrogenase never attached firmly to the proton pore in the membrane. Interestingly the methanogens do still have a cytoplasmic FeNi hydrogenase (or NiFe in this illustration) passing electrons down FeS wires. Instead of dropping them straight on to ferredoxin to generate reduced Fd2- using the proton gradient of the cell wall conducted through a membrane pore (as per proto Ech), pairs of electrons are split at an FAD. Half do the up hill job of generating Fd2- and the other half tumble down hill to the easy target of heterodisulphide. Energy from the latter down hill reaction is used to allow the up hill Fd reduction and gets you out of the need for a membrane proton gradient. I do wonder if this is the same FeNi hydrogenase of proto Ech but here diverted to electron bifurcation. This is from Buckel and Thauer's fantastic paper Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation:

That's what happens today. What might have been the core process when metabolism was less refined? Here's a scheme with FeNi hydrogenase (in Hdr, heterodisulphide reductase) using CH3-SH as the electron acceptor for a crude version of electron bifurcation:

Under circumstances of freely available CH3-SH there is no need to conserve sulphur.
The Ni is shown associated with the enzyme which generates all of the biological methane ever produced on earth, methyl coenzyme-M reductase. Nowadays the Ni is bound in the lovely and highly complex coenzyme F430 (an interesting read if you have access):

Here it is in the step producing methane:

Sulphur is no longer a disposable commodity and it is recycled via CoM as a loop in combination with another sulphydryl based coenzyme, CoB.

CoM provides the -CH3 and CoB provides the -H to generate methane. The reaction joins the two coenzymes together to give the mixed, disulphide bridged, heterodisulphide. This is the modern electron acceptor in the Hdr electron bifurcating hydrogenase. It actually accepts a pair of electrons to give CoM-SH and CoB-SH:

The CoM-SH is regenerated to CH3-S-CoM by the methyltransferase shifting a -CH3 from CH3-H4MPT. Ultimately the energetics of the cell is determined by the availability of CH3-SH analogue CH3-S-CoM controlling electron bifurcation at Hdr:

Next let's take out the electron bifurcation system having established a central role for CH3-S-CoM to energetics control and add in the proton port being used to generate Fd2- using the vent H+ gradient:

And add in the antiporter for Na+ ions:

At this point there is some benefit to converting a protein-translocase to a Na+ driven ATP synthase. Consider that it is Na+ ions that are stabilising the membrane section of the translocase so it seems logical to accept a sudden Na+ gradient as the force pushing inwards to reverse the translocase to generate ATP. So we need the A1-A0 ATP synthase adding in to the diagram:
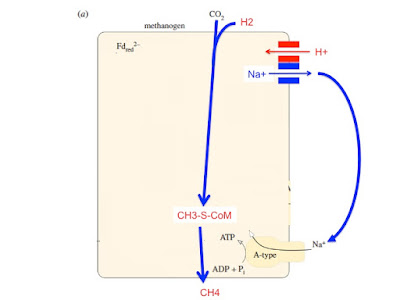
Now a proton gradient is once again driving a Na+ coupled ATP synthase and all is hunky dory for the cell. Excess ATP synthesis can be regulated by CH3-S-CoM inhibiting Na+ antiporting:

When the vent proton gradient fails all that is needed is for CH3-H4MPT to take over from the vent proton gradient. The transfer of the methyl group from H4MPT to CoM is exergonic and is used to drive a conformation change in B12 which ejects the Na+ ion. Lots of detail from Thauer again at The Na‡-translocating methyltransferase complex from methanogenic archaea.

Which simplifies to this:

Giving a speculative journey leading to how we might have ended up here:

In modern methanogenic archaea Na+ energetics have been carried forward to today. Protons still look like an add on to me. Of course the question posed from here is how similar is the Na+/H+ antiporter of the methanogens to the NouH and NuoL combination in the bacteria, incorporated in to the base of complex I.
Not surprisingly I haven't found anyone crazy enough to float this idea. This is what Thauer has to say about Na+ translocating pores and aspartic acid within the pore channel:
"The second reason for the proposal is that only MtrE has a transmembrane helix with an aspartate residue (Fig. 1), the sequence of this helix in the MtrE subunit from all methanogens being highly conserved: 168-IWGITIGAIGSSTGDVHYGAER-191. An aspartate residue in a transmembrane helix has been shown to be essential for sodium ion translocation as catalyzed by the L-subunit of oxaloacetate decarboxylase from Klebsiella pneumoniae [62]. An aspartate residue is also conserved in the transmembrane helix of the sodium ion-translocating glutaconyl-CoA decarboxylase from Acidaminococcus fermentans and of the sodium ion-translocating methylmalonyl-CoA decarboxylase from Veillonella parva and Propionigenium modestum [63]".
Here is NuoH from complex I with the aspartic acid at D213 picked out in red:

Which rather implies that NuoH, rather than NuoL, was the Na+ part of the antiporter, assuming the membrane portions of methyltransferase and Ech derivatives are distant relatives of the same ancestral protein...
Peter

This has CH3-SH (used at two points in the process) driving acetate formation for cell carbon generation. We know this works because Huber and Wächtershäuser demonstrated the abiotic generation of activated acetate from CO and CH3-SH in the presence of an FeS/NiS slurry. No enzymes, no cofactors, no structure. Energetically, it works. I now want to speculate wildly about other uses of CH3-SH in the development of methanogens and the evolution of methyltranseferase, the archaeal alternative to Ech. Let's get rid of the carbon fixation doodles. The location of CH3-SH might need to change as ideas develop:

In the background to the following speculation we still have a FeNi hydrogenase "visiting" a proton channeling pore to generate reduced ferredoxin using a localised low pH region just inside the cell. In the archaea I'm assuming there is no preformed Na+ pump because the protein translocating precursor never jammed up so never pumped Na+ ions. There was no drop in intracellular Na+ and the FeNi hydrogenase never attached firmly to the proton pore in the membrane. Interestingly the methanogens do still have a cytoplasmic FeNi hydrogenase (or NiFe in this illustration) passing electrons down FeS wires. Instead of dropping them straight on to ferredoxin to generate reduced Fd2- using the proton gradient of the cell wall conducted through a membrane pore (as per proto Ech), pairs of electrons are split at an FAD. Half do the up hill job of generating Fd2- and the other half tumble down hill to the easy target of heterodisulphide. Energy from the latter down hill reaction is used to allow the up hill Fd reduction and gets you out of the need for a membrane proton gradient. I do wonder if this is the same FeNi hydrogenase of proto Ech but here diverted to electron bifurcation. This is from Buckel and Thauer's fantastic paper Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation:

That's what happens today. What might have been the core process when metabolism was less refined? Here's a scheme with FeNi hydrogenase (in Hdr, heterodisulphide reductase) using CH3-SH as the electron acceptor for a crude version of electron bifurcation:

Under circumstances of freely available CH3-SH there is no need to conserve sulphur.
The Ni is shown associated with the enzyme which generates all of the biological methane ever produced on earth, methyl coenzyme-M reductase. Nowadays the Ni is bound in the lovely and highly complex coenzyme F430 (an interesting read if you have access):

Here it is in the step producing methane:

Sulphur is no longer a disposable commodity and it is recycled via CoM as a loop in combination with another sulphydryl based coenzyme, CoB.

CoM provides the -CH3 and CoB provides the -H to generate methane. The reaction joins the two coenzymes together to give the mixed, disulphide bridged, heterodisulphide. This is the modern electron acceptor in the Hdr electron bifurcating hydrogenase. It actually accepts a pair of electrons to give CoM-SH and CoB-SH:

The CoM-SH is regenerated to CH3-S-CoM by the methyltransferase shifting a -CH3 from CH3-H4MPT. Ultimately the energetics of the cell is determined by the availability of CH3-SH analogue CH3-S-CoM controlling electron bifurcation at Hdr:

Next let's take out the electron bifurcation system having established a central role for CH3-S-CoM to energetics control and add in the proton port being used to generate Fd2- using the vent H+ gradient:

And add in the antiporter for Na+ ions:

At this point there is some benefit to converting a protein-translocase to a Na+ driven ATP synthase. Consider that it is Na+ ions that are stabilising the membrane section of the translocase so it seems logical to accept a sudden Na+ gradient as the force pushing inwards to reverse the translocase to generate ATP. So we need the A1-A0 ATP synthase adding in to the diagram:

Now a proton gradient is once again driving a Na+ coupled ATP synthase and all is hunky dory for the cell. Excess ATP synthesis can be regulated by CH3-S-CoM inhibiting Na+ antiporting:

When the vent proton gradient fails all that is needed is for CH3-H4MPT to take over from the vent proton gradient. The transfer of the methyl group from H4MPT to CoM is exergonic and is used to drive a conformation change in B12 which ejects the Na+ ion. Lots of detail from Thauer again at The Na‡-translocating methyltransferase complex from methanogenic archaea.

Which simplifies to this:

Giving a speculative journey leading to how we might have ended up here:

In modern methanogenic archaea Na+ energetics have been carried forward to today. Protons still look like an add on to me. Of course the question posed from here is how similar is the Na+/H+ antiporter of the methanogens to the NouH and NuoL combination in the bacteria, incorporated in to the base of complex I.
Not surprisingly I haven't found anyone crazy enough to float this idea. This is what Thauer has to say about Na+ translocating pores and aspartic acid within the pore channel:
"The second reason for the proposal is that only MtrE has a transmembrane helix with an aspartate residue (Fig. 1), the sequence of this helix in the MtrE subunit from all methanogens being highly conserved: 168-IWGITIGAIGSSTGDVHYGAER-191. An aspartate residue in a transmembrane helix has been shown to be essential for sodium ion translocation as catalyzed by the L-subunit of oxaloacetate decarboxylase from Klebsiella pneumoniae [62]. An aspartate residue is also conserved in the transmembrane helix of the sodium ion-translocating glutaconyl-CoA decarboxylase from Acidaminococcus fermentans and of the sodium ion-translocating methylmalonyl-CoA decarboxylase from Veillonella parva and Propionigenium modestum [63]".
Here is NuoH from complex I with the aspartic acid at D213 picked out in red:

Which rather implies that NuoH, rather than NuoL, was the Na+ part of the antiporter, assuming the membrane portions of methyltransferase and Ech derivatives are distant relatives of the same ancestral protein...
Peter
Cholesterol reflectivity at 400nm
I'm interested in paleobiology at the moment, readers might have noticed. Mostly how we might have gotten to where we are now without a Sky Pixie. The ancient past is fascinating. Not only single carbon chemistry under far from equilibrium conditions but big stuff like dinosaurs and cardiologists.
People love dinosaurs. If it's not LDLc levels, it's particle counts or apoB numbers. Am I going to die (yes!)? Will it be from CDV or cancer? Let us examine the entrails of a lipid hypothesis which lies eviscerated here before us. Messy.
How many apoBs can I see???? Do they have purple spots (i.e. does the apoB particle under the microscope have focal regions which are hyper-reflective to photons of 400nm wavelength when irradiated by full spectrum bullshit?).
I think we have to think have to respect the views of the president of the ACC. ACC is the American College of Cardiologists. These folks are wildly intelligent, free thinking, adventurous, swashbuckling promoters of new ideas. The newest idea on view from the president of the ACC is this one:
‘Some prominent cardiologists have questioned the 2013 guidelines, but the ACC and AHA have shown little appetite to return to LDL targets. “LDL may or may not correlate to cardiovascular outcomes,” Dr. Kim Allan Williams, president of the ACC, told Reuters last week1.’
Malcolm Kendrick has an excellent post up on this quote. But let's just say it again, it really is so good:
“LDL may or may not correlate to cardiovascular outcomes”
By whom?
Dr. Kim Allan Williams.
Who is?
President of the ACC.
Which is?
The American College of Cardiologists.
Of course, the statement does leave open the possibility that cardiovascular outcomes MAY correlate to LDL levels. More likely not.
Peter
People love dinosaurs. If it's not LDLc levels, it's particle counts or apoB numbers. Am I going to die (yes!)? Will it be from CDV or cancer? Let us examine the entrails of a lipid hypothesis which lies eviscerated here before us. Messy.
How many apoBs can I see???? Do they have purple spots (i.e. does the apoB particle under the microscope have focal regions which are hyper-reflective to photons of 400nm wavelength when irradiated by full spectrum bullshit?).
I think we have to think have to respect the views of the president of the ACC. ACC is the American College of Cardiologists. These folks are wildly intelligent, free thinking, adventurous, swashbuckling promoters of new ideas. The newest idea on view from the president of the ACC is this one:
‘Some prominent cardiologists have questioned the 2013 guidelines, but the ACC and AHA have shown little appetite to return to LDL targets. “LDL may or may not correlate to cardiovascular outcomes,” Dr. Kim Allan Williams, president of the ACC, told Reuters last week1.’
Malcolm Kendrick has an excellent post up on this quote. But let's just say it again, it really is so good:
“LDL may or may not correlate to cardiovascular outcomes”
By whom?
Dr. Kim Allan Williams.
Who is?
President of the ACC.
Which is?
The American College of Cardiologists.
Of course, the statement does leave open the possibility that cardiovascular outcomes MAY correlate to LDL levels. More likely not.
Peter
Sunday, August 16, 2015
When low fat wins
I think I’ve said before, I’m a calories-in, calories-out sort of person. Nothing as simple as losing a kilo of stored fat for every 9000kcal deficit in dietary consumption (or increase in exercise) of course. This is, as we all know, incorrect and of absolutely no use whatsoever in planning an attempt to generate a normal bodyweight.
I am also very aware that, outside a metabolic ward, it is very difficult to even approximately assess a given person’s level of energy output, assuming they are fully compliant with a fixed composition, fixed caloric input. Which they probably aren’t, much of the time. But calories out will be reflected in many more outputs than can achieved by the limited exercise opportunities afforded during the restrictions of an in-patient metabolic ward study.
As Hall commented, the repeated superiority of weight loss by carbohydrate restriction has always been achieved in outpatient studies, never in tightly controlled metabolic ward studies. He didn’t mention that the advantages from carbohydrate restriction were always achieved under calorically unrestricted circumstances in comparison to calorically restricted alternative diets. I’ll just mention that now.
We can also say, with some degree of certainly, that under very tightly controlled in-patient conditions, extreme dietary fat restriction (less than 8% of calories) produced more stored fat loss than a modest reduction in carbohydrate restriction, provided both groups are rigidly forced to cut calories and to limit their exercise to a specified level, for six days. My own feeling is that this is probably true under the circumstances of the study. It provides a very small piece of data of very limited application to the real world. As Hall writes:
"Translation of our results to real-world weight-loss diets for treatment of obesity is limited since the experimental design [and model simulations] relied on strict control of food intake, which is unrealistic in free-living individuals".
I am very lucky.
I don’t live in a metabolic ward. If the weather is cool outside and I feel warm enough to not need my jacket when I let the chickens out in the morning, so be it. If both the air and water temp are 4degC but there is a four foot swell with clean waves shaping up in First Bay I’m going to be thinking about the roof rack, my playboating kayak and my drysuit. I’m guessing that there are few near freezing surf opportunities in a metabolic ward.
People might also be aware that I rather like fatty acids, especially free fatty acids. These uncouple respiration. Uncoupled respiration, at the mitochondrial level, generates heat and so increases metabolic rate. For people with a heathy interest in cold water kayaking, this has its advantages. Sitting in a metabolic ward eating 140g of carbohydrate per day is not going to increase my free fatty acids to a level were uncoupling is going to feature in my metabolism.
The average person with a BMI of 35 is probably going to be running their metabolism under the Crabtree effect. Increased dependence on glycolysis at the expense of reduced utilisation of mitochondrial beta oxidation. While it is quite possible to immediately and markedly increase fatty acid oxidation, there are limits set by how many mitochondria a given cell possesses. The moderate carbohydrate group did increase their fatty acid oxidation, but not enough to compensate for the loss of carbohydrate available from the diet. This is perhaps most clearly seen in Table 3 where a significant drop in sleeping metabolic rate occurred in the moderate carb group and an actual increase was seen in the very fat restricted group. It's probably why the moderate carbohydrate group had a suggestion of increased protein degradation compared to the very low fat group.
Even thought it was nearly 15 years ago, I can still recall Atkins Flu™ as I switched to deeply ketogenic eating from a fairly reasonable modern diet. The "flu" lasted about 6 days (apologies for exactifying my recall to fit with the study duration!) with further acclimatisation over the next few months. There should be no such problem with increasing glycolysis in the very low fat group if you are already running your metabolism on starch combined with HFCS. It is no major problem to up regulate carbohydrate metabolism when it is your normal metabolic fuel source.
So for the first six days of an enforced, calorically restricted, non-ketogenic diet, cutting fat rules provided the restriction is very, very extreme. Do this long term and you will, of course, fail.
Peter
I am also very aware that, outside a metabolic ward, it is very difficult to even approximately assess a given person’s level of energy output, assuming they are fully compliant with a fixed composition, fixed caloric input. Which they probably aren’t, much of the time. But calories out will be reflected in many more outputs than can achieved by the limited exercise opportunities afforded during the restrictions of an in-patient metabolic ward study.
As Hall commented, the repeated superiority of weight loss by carbohydrate restriction has always been achieved in outpatient studies, never in tightly controlled metabolic ward studies. He didn’t mention that the advantages from carbohydrate restriction were always achieved under calorically unrestricted circumstances in comparison to calorically restricted alternative diets. I’ll just mention that now.
We can also say, with some degree of certainly, that under very tightly controlled in-patient conditions, extreme dietary fat restriction (less than 8% of calories) produced more stored fat loss than a modest reduction in carbohydrate restriction, provided both groups are rigidly forced to cut calories and to limit their exercise to a specified level, for six days. My own feeling is that this is probably true under the circumstances of the study. It provides a very small piece of data of very limited application to the real world. As Hall writes:
"Translation of our results to real-world weight-loss diets for treatment of obesity is limited since the experimental design [and model simulations] relied on strict control of food intake, which is unrealistic in free-living individuals".
I am very lucky.
I don’t live in a metabolic ward. If the weather is cool outside and I feel warm enough to not need my jacket when I let the chickens out in the morning, so be it. If both the air and water temp are 4degC but there is a four foot swell with clean waves shaping up in First Bay I’m going to be thinking about the roof rack, my playboating kayak and my drysuit. I’m guessing that there are few near freezing surf opportunities in a metabolic ward.
People might also be aware that I rather like fatty acids, especially free fatty acids. These uncouple respiration. Uncoupled respiration, at the mitochondrial level, generates heat and so increases metabolic rate. For people with a heathy interest in cold water kayaking, this has its advantages. Sitting in a metabolic ward eating 140g of carbohydrate per day is not going to increase my free fatty acids to a level were uncoupling is going to feature in my metabolism.
The average person with a BMI of 35 is probably going to be running their metabolism under the Crabtree effect. Increased dependence on glycolysis at the expense of reduced utilisation of mitochondrial beta oxidation. While it is quite possible to immediately and markedly increase fatty acid oxidation, there are limits set by how many mitochondria a given cell possesses. The moderate carbohydrate group did increase their fatty acid oxidation, but not enough to compensate for the loss of carbohydrate available from the diet. This is perhaps most clearly seen in Table 3 where a significant drop in sleeping metabolic rate occurred in the moderate carb group and an actual increase was seen in the very fat restricted group. It's probably why the moderate carbohydrate group had a suggestion of increased protein degradation compared to the very low fat group.
Even thought it was nearly 15 years ago, I can still recall Atkins Flu™ as I switched to deeply ketogenic eating from a fairly reasonable modern diet. The "flu" lasted about 6 days (apologies for exactifying my recall to fit with the study duration!) with further acclimatisation over the next few months. There should be no such problem with increasing glycolysis in the very low fat group if you are already running your metabolism on starch combined with HFCS. It is no major problem to up regulate carbohydrate metabolism when it is your normal metabolic fuel source.
So for the first six days of an enforced, calorically restricted, non-ketogenic diet, cutting fat rules provided the restriction is very, very extreme. Do this long term and you will, of course, fail.
Peter
Subscribe to:
Posts (Atom)
