Just starting to find a little time to post. This seems like a worthwhile snippet as a follow on to the anacetrapib post, easier to put up than the cooking epics on insulin and the liver, parts two onwards........
Ok, the usual recap:
First there was cholesterol. It was bad, life was simple.
Then came Good cholesterol, HDL battling the Bad cholesterol, LDL.
Then there was Good LDL, large buoyant battling with Really Bad LDL, small dense LDL, sdLDL.
Not only that but native LDL appears to be harmless, it's only oxidised LDL which is the killer, oxLDL.
So the evil sdLDL is only really evil because it is more easily oxidised than fluffier LDL. Maybe, but in general I tend to have glazed over by now, befuddled by the blur of the moving goal posts.
But just occasionally something does grab my attention, especially if it markedly deepens the hole being dug for itself by the lipid hypothesis, like anacetrapib.
It was thanks to Dr Davis that I grasped the concept of CETP inhibitors as eliminators of sdLDL. That's what they are. If you believe in the lipid hypothesis it must be pretty interesting to have a drug which virtually eliminates sdLDL while increasing the body count for cardiovascular deaths, even if 4 dead out of 808 vs 1 dead out of 804 does not reach statistical significance.
But the real gem from Dr D was the finding that anthocyanins are CETP inhibitors. You no longer need to sign up for the next anacetrapib trial to die of a heart attack in the cause of the lipid hypothesis. You can buy a do-it-yourself CETP inhibitor in the form of a purple plant dye.
EDIT pre posting: It always amazes me that someone as perceptive on blood glucose, and indirectly on blood insulin, as Dr Davis can still believe the lipid hypothesis. Really believe. Fascinating.
There now, we all know plants are all natural, healthy and safe. Perhaps that includes recreational plants like Deadly Nightshade, Nux Vomica and Henbane. As an alternative to anacetrapib you can only hope the anthocyanins don't work!
If they do work at least you might have the consolation that you died with cracking lipids.
Peter
PS, taken from here on flavonoids, discussed here:
"However, a similar decrease in protein oxidation [on flavonoid elimination], in 8-oxo-dG excretion and in the increased resistance of plasma lipoproteins to oxidation in the present study points to a more general relief of oxidative stress after depletion of flavonoid- and ascorbate-rich fruits and vegetables from the diet, contrary to common beliefs."
Want oxidative damage? Munch those flavonoids; and the anthocyanin flavonoids come with the added toxicity of CETP inhibition. Mmmmmm, purple fruit!
Tuesday, December 28, 2010
Sunday, December 12, 2010
High fat diet and fertility

Not likely to re establish posting in the next few days (!!!!!) but she is lovely!
Peter
Saturday, November 27, 2010
The anacetrapib giggle
Stan has a link up to the full text but here is the fun table:

My summary is that a much larger study is desperately needed to confirm that the 300% increase found in cardiovascular mortality is a direct effect of anacetrapib. An even larger study is also urgently required to demonstrate statistical significance for the more modest increase in all cause mortality caused by this drug.
Volunteers should join the queue marked "idiot", unless they are certain that they will get in to the placebo group.
Peter
Still no blogging but this was too fun to skip.

My summary is that a much larger study is desperately needed to confirm that the 300% increase found in cardiovascular mortality is a direct effect of anacetrapib. An even larger study is also urgently required to demonstrate statistical significance for the more modest increase in all cause mortality caused by this drug.
Volunteers should join the queue marked "idiot", unless they are certain that they will get in to the placebo group.
Peter
Still no blogging but this was too fun to skip.
Sunday, October 31, 2010
Hepatic extraction of fructose
EDIT on 2nd March 2011. This post is incorrect! I'll leave it up as a reminder to myself to check all facts, even when net access is very limited. The pancreas monitors enteric glucose hormonally, not by direct access to the portal vein blood flow. It gets as much or as little fructose exposure as any organ than the liver. Mea culpa. The links are good, so the post has some use still. Thanks to Kurt for catching this one for me. END EDIT
OK, still no posting except for this brief note which is only delivered because the clocks changed, Daniel had a disturbed night and we both have been wake for several hours. Another heavy clinical week to come as of tomorrow... I publish the non-viagra comments on older posts by a brief mouse click but still don't get time to comment back and that will probably apply to this post too. That's just how it is at the moment. Tee hee, probably means there are a ton(ne) of typos in this post too!
Over the last few months I've tried to keep up with my favourite blogs (difficult because work blocks access to all blogs). Many people commented on the fructose and cancer article via Reuters. I'd just like to stick a few observations down about it and about hepatic fructose extraction.
Don over at Primal Wisdom has a nice post discussing the subject and its possible implications. But does fructose feed cancer in vivo? Does it even get to any cancer cells outside the liver and gut?
This led to a follow on post about hepatic fructose extraction, with a nice paper from Japan in Diabetes Care cited. It's unfortunate that ref 11 and ref 13 from this article are both unavailable, even in abstract form. One title specifies investigation of splanchnic blood levels of fructose, which is a very vague term but might include portal vein from gut to liver and might just, if we were lucky, include hepatic vein concentrations, which would allow us to see hepatic extraction rate and systemic penetration.
This would be nice as the second reference's title only seems to specify the role of the liver in the mop-up following intravenous fructose administration. Obviously intravenous fructose bypasses hepatic extraction, so it might not say too much about dietary fructose penetration in to the systemic circulation.
When we look at the micro molar concentrations of fructose (as opposed to milli molar for glucose) in serum as measured in DC article we are looking at venous samples. If the fructose in these samples has come from the diet in the gut it will have been fructose extracted by the liver, then pumped around the body, been fructose extracted by the tissues and only then finally arrived at the sampling needle. It looks like an open question how much fructose comes past the liver and hits the tissues themselves, to feed cancer cells.
You might get more information by measuring the arterial concentration of fructose, as opposed to the venous concentration. Arterial concentration is the hepatic vein fructose diluted in the the full venous return/cardiac output. This would give an indication of hepatic fructose passage without the complication of tissue extraction. But you can't have what's not in the paper and arterial blood samples are a little harder to obtain than venous samples!
EDIT: Cynthia found the abstract to the rat paper. Hepatic extraction is around 50-70%, this dilutes in the venous return but there is still a significant surge through the systemic circulation. This is particularly interesting as humans do metabolise fructose in their muscles, which might just have something to do with systemic as well as hepatic insulin resistance from fructose. I wish I had the time to follow these leads. Abstract text in the first comment. Ta Cynthia. END EDIT
The DC paper does suggest, among several explanations, that the higher venous fructose in diabetics might come from glucose via the polyol pathway. Glucose to sorbitol, sorbitol to fructose. This endogenously generated fructose then drops in to the venous system and gets sampled before going off to liver and/or muscles for metabolism.
So there are a lot of "if"s, "but"s and "maybe"s.
However, the cancer feeding effect of fructose was noted in pancreatic cells. Pancreatic cells sit in the portal vein to monitor gut glucose absorption. They also get hit by the full load of fructose arriving after a couple of cans of soda. It doesn't matter how much fructose the liver extracts if you happen to be a pancreatic cell sitting in the portal blood flow.
You get nuked.
If you are a pancreatic cancer cell you get fed.
Peter
BTW NAFLD has it's parallel in non alcoholic fatty pancreatic disease. Both go from normal through fatty infiltration to chronic inflammation to scarring to neoplasia. Both organs sit in the portal venous drainage from the gut. Another few posts there but...
OK, still no posting except for this brief note which is only delivered because the clocks changed, Daniel had a disturbed night and we both have been wake for several hours. Another heavy clinical week to come as of tomorrow... I publish the non-viagra comments on older posts by a brief mouse click but still don't get time to comment back and that will probably apply to this post too. That's just how it is at the moment. Tee hee, probably means there are a ton(ne) of typos in this post too!
Over the last few months I've tried to keep up with my favourite blogs (difficult because work blocks access to all blogs). Many people commented on the fructose and cancer article via Reuters. I'd just like to stick a few observations down about it and about hepatic fructose extraction.
Don over at Primal Wisdom has a nice post discussing the subject and its possible implications. But does fructose feed cancer in vivo? Does it even get to any cancer cells outside the liver and gut?
This led to a follow on post about hepatic fructose extraction, with a nice paper from Japan in Diabetes Care cited. It's unfortunate that ref 11 and ref 13 from this article are both unavailable, even in abstract form. One title specifies investigation of splanchnic blood levels of fructose, which is a very vague term but might include portal vein from gut to liver and might just, if we were lucky, include hepatic vein concentrations, which would allow us to see hepatic extraction rate and systemic penetration.
This would be nice as the second reference's title only seems to specify the role of the liver in the mop-up following intravenous fructose administration. Obviously intravenous fructose bypasses hepatic extraction, so it might not say too much about dietary fructose penetration in to the systemic circulation.
When we look at the micro molar concentrations of fructose (as opposed to milli molar for glucose) in serum as measured in DC article we are looking at venous samples. If the fructose in these samples has come from the diet in the gut it will have been fructose extracted by the liver, then pumped around the body, been fructose extracted by the tissues and only then finally arrived at the sampling needle. It looks like an open question how much fructose comes past the liver and hits the tissues themselves, to feed cancer cells.
You might get more information by measuring the arterial concentration of fructose, as opposed to the venous concentration. Arterial concentration is the hepatic vein fructose diluted in the the full venous return/cardiac output. This would give an indication of hepatic fructose passage without the complication of tissue extraction. But you can't have what's not in the paper and arterial blood samples are a little harder to obtain than venous samples!
EDIT: Cynthia found the abstract to the rat paper. Hepatic extraction is around 50-70%, this dilutes in the venous return but there is still a significant surge through the systemic circulation. This is particularly interesting as humans do metabolise fructose in their muscles, which might just have something to do with systemic as well as hepatic insulin resistance from fructose. I wish I had the time to follow these leads. Abstract text in the first comment. Ta Cynthia. END EDIT
The DC paper does suggest, among several explanations, that the higher venous fructose in diabetics might come from glucose via the polyol pathway. Glucose to sorbitol, sorbitol to fructose. This endogenously generated fructose then drops in to the venous system and gets sampled before going off to liver and/or muscles for metabolism.
So there are a lot of "if"s, "but"s and "maybe"s.
However, the cancer feeding effect of fructose was noted in pancreatic cells. Pancreatic cells sit in the portal vein to monitor gut glucose absorption. They also get hit by the full load of fructose arriving after a couple of cans of soda. It doesn't matter how much fructose the liver extracts if you happen to be a pancreatic cell sitting in the portal blood flow.
You get nuked.
If you are a pancreatic cancer cell you get fed.
Peter
BTW NAFLD has it's parallel in non alcoholic fatty pancreatic disease. Both go from normal through fatty infiltration to chronic inflammation to scarring to neoplasia. Both organs sit in the portal venous drainage from the gut. Another few posts there but...
Friday, October 15, 2010
A heads up
Just a brief heads up, especially to people who have emailed me off blog, apologies for the near total lack of replies, there really is no net time worth speaking of at the moment. Too busy cutting and stitching lots of stuff and other things! When we get in to a house of our own things will get back to some semblance of normality but that is not looking like happening in the immediate future.
Peter
Among the many links I've not had time to follow there was this link which I did manage to clicked on and couldn't leave alone. Thanks Elizabeth.
“It’s like an epidemic, in the sense that they’re infected with these wrong ideas, and they’re spreading it to other researchers through journals.”
Hee hee, Ioannidis does meme watching...
Peter
Among the many links I've not had time to follow there was this link which I did manage to clicked on and couldn't leave alone. Thanks Elizabeth.
“It’s like an epidemic, in the sense that they’re infected with these wrong ideas, and they’re spreading it to other researchers through journals.”
Hee hee, Ioannidis does meme watching...
Thursday, September 23, 2010
von Gierke's disease
I'm just taking a quick break from packing boxes and trawling through hepatic insulin resistance related to metabolic syndrome because I got (as always) side tracked. By hepatic glycogen storage this time, rather than lipid storage.
You have to giggle about glycogen storage disease type Ia. It may not be much fun if you have it, but at least you are protected against premature cardiovascular disease.
About von Gierke's disease:
"Glycogen storage disease type Ia (GSD-Ia) is characterized by hypercholesterolemia, hypertriglyceridemia, decreased cholesterol in high density lipoprotein and increased cholesterol in low and very low density lipoprotein fractions."
Of course, with lipids like those, you should die of CVD at a very early age. But you don't.
As people may recall my current hypothesis for the cause of premature CVD is that it is triggered by Purple Spotted sdLDL, something which is NEVER measured by lipidologists. This is hardly surprising because I made it up. Taking a lesson from the lipid hypothesis founders there.
So folks with glycogen storage disease type Ia have the worst possible lipid profile you can imagine and no premature CVD.
To explain this paradox (gasp in awe at the explanations) you can look at antioxidants like uric acid or do very clever things with cholesterol efflux mediators or hypothesise about adiponectin. Take your pick.
Guess what. People with glycogen storage disease type Ia are virtually never hyperglycaemic or hyperinsulinaemic. In fact hypoglycaemia can be a serious problem for them. But they don't get heart disease. Funny that.
Oh, and they are told to avoid fructose too (haven't checked why, but it seems like a good idea)... Perhaps I'm wrong about Purple Spotted sdLDL, it could just be that it's made of fructose rather than sucrose!
Peter
You have to giggle about glycogen storage disease type Ia. It may not be much fun if you have it, but at least you are protected against premature cardiovascular disease.
About von Gierke's disease:
"Glycogen storage disease type Ia (GSD-Ia) is characterized by hypercholesterolemia, hypertriglyceridemia, decreased cholesterol in high density lipoprotein and increased cholesterol in low and very low density lipoprotein fractions."
Of course, with lipids like those, you should die of CVD at a very early age. But you don't.
As people may recall my current hypothesis for the cause of premature CVD is that it is triggered by Purple Spotted sdLDL, something which is NEVER measured by lipidologists. This is hardly surprising because I made it up. Taking a lesson from the lipid hypothesis founders there.
So folks with glycogen storage disease type Ia have the worst possible lipid profile you can imagine and no premature CVD.
To explain this paradox (gasp in awe at the explanations) you can look at antioxidants like uric acid or do very clever things with cholesterol efflux mediators or hypothesise about adiponectin. Take your pick.
Guess what. People with glycogen storage disease type Ia are virtually never hyperglycaemic or hyperinsulinaemic. In fact hypoglycaemia can be a serious problem for them. But they don't get heart disease. Funny that.
Oh, and they are told to avoid fructose too (haven't checked why, but it seems like a good idea)... Perhaps I'm wrong about Purple Spotted sdLDL, it could just be that it's made of fructose rather than sucrose!
Peter
Saturday, September 18, 2010
Fathead Supersize Me and Sweden (2)
An addendum to the mouse trans fat fibrosis paper: It's junk. Utter junk. Even the title is wrong. There are NO trans fats in the experiment, NONE what so ever!
The mice were fed on the Surwit Diet. Here's the pdf from Research Diets.
I thought medium chain trans fats were a bit of a strange animal....
So the mice are being fed on fully hydrogenated coconut oil, which has minimal PUFA to begin with and gets hydrogenated to fully saturated, mostly medium chain triglycerides. Plus non hydrogenated soya oil, which is mostly omega 6 PUFA and a little omega 3 PUFA.
The ONLY source of carbohydrate in the diet was sucrose and maltodextrin, which made up 25% of calories. To which they added sucrose in the drinking water, plus added fructose in the drinking water.
THERE ARE NO TRANS FATS IN THIS DIET.
Fructose, beyond human comprehension, with adequate soya oil, is enough to fibrose a mouse's liver. Not even Bill Clinton could eat this much fructose a day for life.
Who scrutineered this paper? Who wrote the title? How much money was wasted? What is happening in the world?
Arghhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhh!
Ouch. Ouch. Ouch.
Ouch.
I'm trying to stop banging my head on the table but (Ouch)......
Peter
The mice were fed on the Surwit Diet. Here's the pdf from Research Diets.
I thought medium chain trans fats were a bit of a strange animal....
So the mice are being fed on fully hydrogenated coconut oil, which has minimal PUFA to begin with and gets hydrogenated to fully saturated, mostly medium chain triglycerides. Plus non hydrogenated soya oil, which is mostly omega 6 PUFA and a little omega 3 PUFA.
The ONLY source of carbohydrate in the diet was sucrose and maltodextrin, which made up 25% of calories. To which they added sucrose in the drinking water, plus added fructose in the drinking water.
THERE ARE NO TRANS FATS IN THIS DIET.
Fructose, beyond human comprehension, with adequate soya oil, is enough to fibrose a mouse's liver. Not even Bill Clinton could eat this much fructose a day for life.
Who scrutineered this paper? Who wrote the title? How much money was wasted? What is happening in the world?
Arghhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhh!
Ouch. Ouch. Ouch.
Ouch.
I'm trying to stop banging my head on the table but (Ouch)......
Peter
Thursday, September 16, 2010
Fathead, Supersize Me and Sweden
I generally ignored this paper in 2008 as it didn't look particularly interesting and seemed mostly about fast food bashing, where the fast food included a large amount of sugar and starch. A bit like a real life Supersize Me, but with genuine average food intakes provided and individual responses in ALT levels, a marker traditionally associated with liver damage, also provided.
If anyone wants to eat 285g of sugar a day then they deserve whatever they have coming to them. What they have coming is an ALT increase which correlates with either carbohydrate or sugar intake by three weeks in to the feast. Not with fat.
I found the lack of association with fat disappointing. Sweden is a country fairly replete with trans fat and a little arithmetic applied to Table 1 (LeenaS describes these as "hidden fats") suggests that trans fat intake went from about 6g/d to 24g/d during the over feeding period of the study. This is not quite at the level of Crisco poisoning but I was still disappointed to see no discernible association with ALT. Ah well, you can't have everything.
Now I remember Tom Naughton nuking himself with trans fats during Fathead to the point of lowering his HDL, but otherwise he developed no suggestion of metabolic syndrome. But then Tom engaged his brain before drinking bucket loads of fructose and desisted from such stupidity. The fructose trick is for anyone with more Spurlockian intelligence.
To go back to Sweden: Gross overfeeding with "fast food" elevates your ALT. This is sort of boring because no one in their right mind is going to eat that much fructose in a month, which sort of defines Spurlock.
But was anyone still awake by the end of Supersize Me? Remember how long it took him to lose weight on his girlfriend's vegan cooking?
Now the question is, does this translate across in to the Swedish ovefeeding group? Well that was answered by a follow on study looking at the participants two and a half years down the road.
They're still fat. On average.
Something breaks in a month of overfeeding with trans fats and sugar. That is fascinating. Now you could argue that the volunteers got the taste for junk food, that their fat cells got stretched, that they were already self selected for being comfortable with gaining weight to take part in the study etc. I'd like to look a little more closely at liver pathology.
Elevated ALT is traditionally assumed to indicate liver damage. But there might be circumstances where you make more ALT in each cell, especially if a lot of amino acid processing is going on, so get benign ALT elevation. No one had a liver biopsy so it was impossible to find out exactly why the ALT went up.
Now, from a pathologist's point of view, a fatty liver is completely reversible. There is nothing permanently damaged in each hepatocyte or in the structure of the liver. Hepatic inflammation is also theoretically reversible. Those old leucocytes can leg it out just as easily as they legged it in.
But fibrosis, that's a different matter. Fibrosis is there to stay. This is at the micro architectural level. We're not talking cirrhosis (yet). No pathologist expects a fibrosed liver to go back to normal. It may adapt, regenerate, keep you alive, yes. But it's not normal. It will never be normal.
I picked up a link to a mouse study in which they fed chow, Super Crisco (medium chain trans fats, what are they?) or Super Crisco plus fructose enriched sucrose via the drinking water. The rest of the diet is not in the abstract so who knows what else they did. But it was the Super Crisco plus fructose/sucrose in the drinking water which made all of the headlines.
Super Crisco appears to be bad for your waistline but may not, on its own, produce the irreversible changes in the liver seen in the mice who combined it with HFCS. As far as you can tell from the abstract.
A diet replete in trans fats and HFCS fibroses your liver.
Translating from the Swedish volunteers and these poisoned mice to our two film directors:
Tom Naughton should be fine with his low fructose high trans fat diet and Spurlock should have aged his liver by a few years (in terns of insulin sensitivity) during his month of self poisoning on trans fats because he combined them with fructose to push his ALT through the roof.
I'll try and put this in to a physiology context when a little more time comes my way.
Peter
BTW: A methodological note from the initial Swedish study which adds the "human element" to the mind set of the "scientists" running it:
"If the subject was not able or willing to ingest the hamburger-based diet at any stage, it was changed to whatever food the participant accepted with the highest priority to achieve the calculated caloric intake and also, if the study subject still found it acceptable, a diet rich in protein and saturated animal fat."
My emphasis.
As always, it's nice when people nail their colours to the mast.
I see from Table 1 in the follow-on study that one man and one woman had actually reduced their weight to below their pre study weight by 2.5 years. I just wonder whether they were the ones who refused the trans fats of the hamburger diet and went with animal fat and protein to source their excess calories. No one is saying.
That human element gets everywhere!
EDIT: See Patrick's notes in the comments about the group leader from the hyperalimentation studies. A different impression from the published papers. There is hope for Sweden. Good.
If anyone wants to eat 285g of sugar a day then they deserve whatever they have coming to them. What they have coming is an ALT increase which correlates with either carbohydrate or sugar intake by three weeks in to the feast. Not with fat.
I found the lack of association with fat disappointing. Sweden is a country fairly replete with trans fat and a little arithmetic applied to Table 1 (LeenaS describes these as "hidden fats") suggests that trans fat intake went from about 6g/d to 24g/d during the over feeding period of the study. This is not quite at the level of Crisco poisoning but I was still disappointed to see no discernible association with ALT. Ah well, you can't have everything.
Now I remember Tom Naughton nuking himself with trans fats during Fathead to the point of lowering his HDL, but otherwise he developed no suggestion of metabolic syndrome. But then Tom engaged his brain before drinking bucket loads of fructose and desisted from such stupidity. The fructose trick is for anyone with more Spurlockian intelligence.
To go back to Sweden: Gross overfeeding with "fast food" elevates your ALT. This is sort of boring because no one in their right mind is going to eat that much fructose in a month, which sort of defines Spurlock.
But was anyone still awake by the end of Supersize Me? Remember how long it took him to lose weight on his girlfriend's vegan cooking?
Now the question is, does this translate across in to the Swedish ovefeeding group? Well that was answered by a follow on study looking at the participants two and a half years down the road.
They're still fat. On average.
Something breaks in a month of overfeeding with trans fats and sugar. That is fascinating. Now you could argue that the volunteers got the taste for junk food, that their fat cells got stretched, that they were already self selected for being comfortable with gaining weight to take part in the study etc. I'd like to look a little more closely at liver pathology.
Elevated ALT is traditionally assumed to indicate liver damage. But there might be circumstances where you make more ALT in each cell, especially if a lot of amino acid processing is going on, so get benign ALT elevation. No one had a liver biopsy so it was impossible to find out exactly why the ALT went up.
Now, from a pathologist's point of view, a fatty liver is completely reversible. There is nothing permanently damaged in each hepatocyte or in the structure of the liver. Hepatic inflammation is also theoretically reversible. Those old leucocytes can leg it out just as easily as they legged it in.
But fibrosis, that's a different matter. Fibrosis is there to stay. This is at the micro architectural level. We're not talking cirrhosis (yet). No pathologist expects a fibrosed liver to go back to normal. It may adapt, regenerate, keep you alive, yes. But it's not normal. It will never be normal.
I picked up a link to a mouse study in which they fed chow, Super Crisco (medium chain trans fats, what are they?) or Super Crisco plus fructose enriched sucrose via the drinking water. The rest of the diet is not in the abstract so who knows what else they did. But it was the Super Crisco plus fructose/sucrose in the drinking water which made all of the headlines.
Super Crisco appears to be bad for your waistline but may not, on its own, produce the irreversible changes in the liver seen in the mice who combined it with HFCS. As far as you can tell from the abstract.
A diet replete in trans fats and HFCS fibroses your liver.
Translating from the Swedish volunteers and these poisoned mice to our two film directors:
Tom Naughton should be fine with his low fructose high trans fat diet and Spurlock should have aged his liver by a few years (in terns of insulin sensitivity) during his month of self poisoning on trans fats because he combined them with fructose to push his ALT through the roof.
I'll try and put this in to a physiology context when a little more time comes my way.
Peter
BTW: A methodological note from the initial Swedish study which adds the "human element" to the mind set of the "scientists" running it:
"If the subject was not able or willing to ingest the hamburger-based diet at any stage, it was changed to whatever food the participant accepted with the highest priority to achieve the calculated caloric intake and also, if the study subject still found it acceptable, a diet rich in protein and saturated animal fat."
My emphasis.
As always, it's nice when people nail their colours to the mast.
I see from Table 1 in the follow-on study that one man and one woman had actually reduced their weight to below their pre study weight by 2.5 years. I just wonder whether they were the ones who refused the trans fats of the hamburger diet and went with animal fat and protein to source their excess calories. No one is saying.
That human element gets everywhere!
EDIT: See Patrick's notes in the comments about the group leader from the hyperalimentation studies. A different impression from the published papers. There is hope for Sweden. Good.
Tuesday, September 14, 2010
Axen, Axen (3) and Hawks
John Hawks put up this excellent quote in his post James Randi on scientists
From Randi, J. 1988. "The detection of fraud and fakery." Cell Mol Life Sci 44:287-288:
"Scientists are very easily deceived. They think logically, extrapolate possibilities from evidence presented, assume (with a good probability of being right) certain aspects of the observed data and draw upon their past experience in coming to decisions. This is to say that they act very much as all humans do, struggling with sensory input to derive new facts from it. But scientists do this with a certain authority and certainty born of their training and discipline. They are thus excellent candidates for being flimflammed by a clever operator who is aware of the fact that scientists seldom bring the human element into account."
Axen and Axen do not do either fraud or fakery. Their data are real. But the human element is essential.
Peter
From Randi, J. 1988. "The detection of fraud and fakery." Cell Mol Life Sci 44:287-288:
"Scientists are very easily deceived. They think logically, extrapolate possibilities from evidence presented, assume (with a good probability of being right) certain aspects of the observed data and draw upon their past experience in coming to decisions. This is to say that they act very much as all humans do, struggling with sensory input to derive new facts from it. But scientists do this with a certain authority and certainty born of their training and discipline. They are thus excellent candidates for being flimflammed by a clever operator who is aware of the fact that scientists seldom bring the human element into account."
Axen and Axen do not do either fraud or fakery. Their data are real. But the human element is essential.
Peter
Wednesday, September 01, 2010
Axen and Axen (2)
OK, the biggest mistakes in A&A's 2006 paper was, in my book, Atkins bashing. There, I am biased. Citing the Atkins Diet specifically and using this citation as the basis for their study design is problematic. I don't know if A&A ever read ref 1 as cited but, believe me, these rats were not on the Atkins Induction or the subsequent Ongoing Weight Loss phases. Generally the Atkins diet involves Food plus artificial sweeteners and some easily avoidable non-foods such as soy flour. And vegetables.
So what did they do? They took a group of lab rats and made them obese with trans fatty acid enriched Crisco as 60% of their calories. They then split the rats in to two groups, one was given 60% of calories as carbohydrate through out. The other group was given 5% carbohydrate for 2 weeks then 15% carbohydrate for a month, à la Atkins. Both groups were moderately energy restricted, dictated by a somewhat random decision protocol.
There was a parallel group eating crapinabag (CIAB) throughout (no Crisco). Glucose tolerance tests, with insulin measured at 20 minutes, were performed at various time points.
What went wrong in 2006?
Things started well with the Crisco rats having higher blood glucose at 10 minutes in to the GTT than the CIAB rats. Insulin levels were a lot higher in the Criso rats 20 minutes in to the test by which time glucose was identical between Crisco and CIAB groups. That's Graph A. Crisco causes insulin resistance.

Things were going reasonably well for Atkins bashing at the end of the two week "Atkins Induction" phase. During GTT the 60% carb group were slightly lower in glucose and this made p<0.05 at 10 minutes. However the cracks are beginning to show. The "Atkins Induction" group had an insulin at 20 minutes in to GTT of 600pM, the 60% carb group needed an insulin of over 900pM to achieve the marginally lower glucose level at this point. The insulin values were, luckily for A&A, not significantly different. Fasting insulin at this point was also lower in the "Atkins Induction" group. A&A were lucky on the p values here too. Here's graph B with that spiked glucose at 10 minutes:

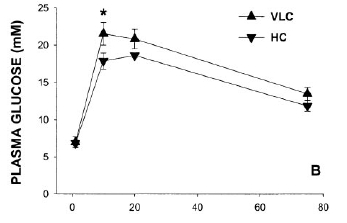
By the end of the experiment at 14 weeks the Atkins Group had been given more (15% of calories) carbohydrate. The GTT at this time point is shown here:

Now you need to get your glasses on for this one. Can you see any difference between the "Atkins Ongoing Weight Loss" (VLC) group and the 60% carbohydrate (HC) weight loss groups? No? Me neither.
Insulin values were slightly better in the 60% carb group but again nothing significant. Both weight loss groups had lower insulin values than the CIAB group! All NS again.
So there we have it: Atkins Ongoing Weight Loss, as interpreted by A&A, gives a GTT curve which is superimposed on the 60% carbohydrate weight loss group. ATKINS is GOOD!
There is a load of bollocks in the discussion about the impaired insulin response in the Atkins group in graph B. To me shifting glucose with a lower insulin level is good, not bad. The spike at 10 minutes is the only saving grace to the funding generating ability of this study.
But graph C is just hysterical.
Okay, A&A are not stupid. They worked out exactly what went wrong in graph C and what was going well in graph B.
So they went out and got more funding to demonstrate CONCLUSIVELY that the Atkins Diet makes you diabetic. They got that funding. These people are good, make no mistake. They got the desired result second time round. How many people get a second chance like this? They published in 2010.
Here is graph a from 2010, directly comparable to graph A from 2006.

Very similar but tidied up in 4 years of refining the model. Or maybe the CIAB has been improved. Anyhoo, same result. Crisco does nasty things to GTT curves.
Next is graph b, which is like graph B above but is after a month rather than 2 weeks and has the on going Crisco group included. The very low carbohydrate group is looking a lot like the Crisco group by now...

But here is the Money Shot in graph c from 2010. Just look at the Crisco curve (HF) and the 5% carbohydrate (VLC) curve. Just look at that fit!

I told you these people were good!
But also go back and look at the blooper graph C from 2006.
So what is going on?
There is a nice pointer in line seven of Table 2, "Soleus TAG". This is the amount of intra myocyte lipid in a typical muscle. It is a marker of how reluctant that muscle is going to be to accept glucose. Two groups have high soleus TAG. The Crisco poisoned (HF) group throughout and the VLC group at 16 weeks.

The explanations for why these two groups have high soleus TAG is likely to be different. Both will, in all certainty, reflect elevated FFAs in the plasma. But the Crisco poisoned group will have elevated FFAs, 24/7, despite 15% of calories as starch. We know from the 2006 blooper that 15% of calories as starch will give a GTT curve in VLC rats which matches the 60% carb group EXACTLY. Not so if you are Crisco poisoned.
The VLC rats on 5% of carbs will have elevated FFAs 24/7 because they would be dead without them. They are on a starvation diet of which only 5% is carbs. Without FFAs they would run their muscles on glucose. They don't get enough glucose per day to do this and still keep their brain alive. Death is not an option.
So the Crisco group has elevated TAG in soleus muscle in the presence of carbohydrate in the diet. It's pathological. The VLC group has elevated TAG in their soleus muscle because they had minimal free glucose available, which is physiological.
BTW either fasting or a brief period without carbohydrate will promptly elevate muscle TAG in humans. It is an utterly normal response to a reduced supply of glucose. The actual signal for muscle insulin resistance is not likley to be the tri acyl glycerol molecules themselves because athletes have bucket loads of this without insulin resistance. More likely is a more ephermeral moiety such as Acyl-CoA molecules or diglycerides which more closely reflect FFA supply. In a GTT the glucose supply is massively supraphysiological. For insulin sensitivity to return to LC muscles it takes time for insulin to spike, insulin to get to adipocytes, adipocytes to respond to insulin, FFA level in blood to drop and FFA derivative level in muscle to drop. It's hardly surprising that the 10 minute glucose peak was higher in the VLC rats during GTT. However, as soon as the muscles clear FFA derivatives they are still geared up to go with glucose, nae problem, nae bother. I'll come on to issues with insulin later. Obviously the Crisco poisoned rats are obese and their adipocytes will have an inability to suppress FFA release in response to insulin. That's how it is if you eat Crisco.
Let's look at the insulin responses. All of the fasting insulin levels were about the same. Obviously the VLC had the lowest insulin and almost certainly the lowest HOMA score although p might still have been > 0.05. By week 16 the insulin response to GTT was interesting.
The VLC rats mimicked the Crisco (HF) group's glucose curve. But they did it with just 1.79ng/ml of insulin. The Crisco rats needed 2.93ng/ml of insulin (p<0.05). The lower curve with open diamonds is the 60% carb group. The curve looks good until you realise that these rats needed as much insulin as the Crisco rats to achieve this beautiful curve, nearly twice that in the VLC group (2.95ng/ml vs 1.79ng/ml, p<0.05).
So which rats are the most insulin sensitive? Not the Crisco rats. I'll accept that. Just say no to Crisco... It is completely arguable between the VCL and 60% carb group.
BUT. What would have happened if the VLC group had produced the same insulin response as the 60% carb group? Impossible, scream Axen and Axen. The VCL group have a blunted insulin response. It makes them well on the road to diabetes, metabolic syndrome, blindness, dialysis, we need the funding...
Calm down Peter, bit OTT there!
Except metabolic syndrome is characterised by elevated insulin, not depressed insulin. Duh.
I have to thank Helen who placed a comment on another post. She pointed out that glucokinase in the pancreas, the enzyme which the pancreas uses to sense glucose in the portal blood, is down regulated in response to carbohydrate restriction. Oh.
It is, err, up regulated in carbohydrate surplus.
This is what "bit" Axen and Axen in 2006. It looks like 15% of calories as carbohydrate in a VLC non-Crisco situation is adequate (on a high protein background) to allow pancreatic insulin secretion in response to glucose to become identical to that produced by rats on a 60% carbohydrate diet. Muscle TAG and associated molecules will drop too. Hence the overlay of the GTT curves in 2006.
Let us assume, very reasonably, that the VLC rats in 2010, on a 5% carb, energy restricted diet, are not expecting to deal with hyperglycaemia any time soon. They down regulate glucokinase production. Then some joker injects 1g/kg of glucose in to their peritoneal cavity. No one up regulates their glucokinase in 10 minutes, not even Super Rat*. Insulin response is blunted. Hyperglycaemia results.
*Actually Super Rat could do this but she is always busy saving the planet (again) and doesn't have time to help out here.
What would have happened with a few days carb loading in the VLC group before the GTT? Well, we (that "we" includes A&A) know the answer to this from 2006. Did you really think A&A are stupid? How many times do I have to point out that these people are good. Very, very good. They know that to get a "bad" result for VLC you must NOT increase carbs pre glucose load in a GTT.
Does anyone think that neither Axen nor Axen has heard of glucokinase? That would mean they're stupid. They're not, they know that if they allowed 15% carbs for a few days the VLC group would overlay the curve of the 60% carb group. For crying out loud, they published the damned curves themselves!
No. The effect of increased carbs on a VLC is not "unclear" (their word). It adjusts pancreatic insulin secretion to deal with carbs when carbs form a significant part of the diet. That's called physiology!
In summary:
Do A&A have a paradigm to support, a mortgage or two to pay, a living to make, careers to develop?
A few fatties getting injured is of no concern, provided the models can be adjusted to keep the funding coming through.
Will people edging towards type 2 diabetes get injured by a very low carbohydrate diet or will they be injured by A&A's funding success? What if they eat low fat high carbohydrate in the real world? What is hunger?
You decide. Then go eat some fat.
Peter
But not Crisco. Just say no....
So what did they do? They took a group of lab rats and made them obese with trans fatty acid enriched Crisco as 60% of their calories. They then split the rats in to two groups, one was given 60% of calories as carbohydrate through out. The other group was given 5% carbohydrate for 2 weeks then 15% carbohydrate for a month, à la Atkins. Both groups were moderately energy restricted, dictated by a somewhat random decision protocol.
There was a parallel group eating crapinabag (CIAB) throughout (no Crisco). Glucose tolerance tests, with insulin measured at 20 minutes, were performed at various time points.
What went wrong in 2006?
Things started well with the Crisco rats having higher blood glucose at 10 minutes in to the GTT than the CIAB rats. Insulin levels were a lot higher in the Criso rats 20 minutes in to the test by which time glucose was identical between Crisco and CIAB groups. That's Graph A. Crisco causes insulin resistance.

Things were going reasonably well for Atkins bashing at the end of the two week "Atkins Induction" phase. During GTT the 60% carb group were slightly lower in glucose and this made p<0.05 at 10 minutes. However the cracks are beginning to show. The "Atkins Induction" group had an insulin at 20 minutes in to GTT of 600pM, the 60% carb group needed an insulin of over 900pM to achieve the marginally lower glucose level at this point. The insulin values were, luckily for A&A, not significantly different. Fasting insulin at this point was also lower in the "Atkins Induction" group. A&A were lucky on the p values here too. Here's graph B with that spiked glucose at 10 minutes:
By the end of the experiment at 14 weeks the Atkins Group had been given more (15% of calories) carbohydrate. The GTT at this time point is shown here:

Now you need to get your glasses on for this one. Can you see any difference between the "Atkins Ongoing Weight Loss" (VLC) group and the 60% carbohydrate (HC) weight loss groups? No? Me neither.
Insulin values were slightly better in the 60% carb group but again nothing significant. Both weight loss groups had lower insulin values than the CIAB group! All NS again.
So there we have it: Atkins Ongoing Weight Loss, as interpreted by A&A, gives a GTT curve which is superimposed on the 60% carbohydrate weight loss group. ATKINS is GOOD!
There is a load of bollocks in the discussion about the impaired insulin response in the Atkins group in graph B. To me shifting glucose with a lower insulin level is good, not bad. The spike at 10 minutes is the only saving grace to the funding generating ability of this study.
But graph C is just hysterical.
Okay, A&A are not stupid. They worked out exactly what went wrong in graph C and what was going well in graph B.
So they went out and got more funding to demonstrate CONCLUSIVELY that the Atkins Diet makes you diabetic. They got that funding. These people are good, make no mistake. They got the desired result second time round. How many people get a second chance like this? They published in 2010.
Here is graph a from 2010, directly comparable to graph A from 2006.

Very similar but tidied up in 4 years of refining the model. Or maybe the CIAB has been improved. Anyhoo, same result. Crisco does nasty things to GTT curves.
Next is graph b, which is like graph B above but is after a month rather than 2 weeks and has the on going Crisco group included. The very low carbohydrate group is looking a lot like the Crisco group by now...

But here is the Money Shot in graph c from 2010. Just look at the Crisco curve (HF) and the 5% carbohydrate (VLC) curve. Just look at that fit!

I told you these people were good!
But also go back and look at the blooper graph C from 2006.
So what is going on?
There is a nice pointer in line seven of Table 2, "Soleus TAG". This is the amount of intra myocyte lipid in a typical muscle. It is a marker of how reluctant that muscle is going to be to accept glucose. Two groups have high soleus TAG. The Crisco poisoned (HF) group throughout and the VLC group at 16 weeks.

The explanations for why these two groups have high soleus TAG is likely to be different. Both will, in all certainty, reflect elevated FFAs in the plasma. But the Crisco poisoned group will have elevated FFAs, 24/7, despite 15% of calories as starch. We know from the 2006 blooper that 15% of calories as starch will give a GTT curve in VLC rats which matches the 60% carb group EXACTLY. Not so if you are Crisco poisoned.
The VLC rats on 5% of carbs will have elevated FFAs 24/7 because they would be dead without them. They are on a starvation diet of which only 5% is carbs. Without FFAs they would run their muscles on glucose. They don't get enough glucose per day to do this and still keep their brain alive. Death is not an option.
So the Crisco group has elevated TAG in soleus muscle in the presence of carbohydrate in the diet. It's pathological. The VLC group has elevated TAG in their soleus muscle because they had minimal free glucose available, which is physiological.
BTW either fasting or a brief period without carbohydrate will promptly elevate muscle TAG in humans. It is an utterly normal response to a reduced supply of glucose. The actual signal for muscle insulin resistance is not likley to be the tri acyl glycerol molecules themselves because athletes have bucket loads of this without insulin resistance. More likely is a more ephermeral moiety such as Acyl-CoA molecules or diglycerides which more closely reflect FFA supply. In a GTT the glucose supply is massively supraphysiological. For insulin sensitivity to return to LC muscles it takes time for insulin to spike, insulin to get to adipocytes, adipocytes to respond to insulin, FFA level in blood to drop and FFA derivative level in muscle to drop. It's hardly surprising that the 10 minute glucose peak was higher in the VLC rats during GTT. However, as soon as the muscles clear FFA derivatives they are still geared up to go with glucose, nae problem, nae bother. I'll come on to issues with insulin later. Obviously the Crisco poisoned rats are obese and their adipocytes will have an inability to suppress FFA release in response to insulin. That's how it is if you eat Crisco.
Let's look at the insulin responses. All of the fasting insulin levels were about the same. Obviously the VLC had the lowest insulin and almost certainly the lowest HOMA score although p might still have been > 0.05. By week 16 the insulin response to GTT was interesting.
The VLC rats mimicked the Crisco (HF) group's glucose curve. But they did it with just 1.79ng/ml of insulin. The Crisco rats needed 2.93ng/ml of insulin (p<0.05). The lower curve with open diamonds is the 60% carb group. The curve looks good until you realise that these rats needed as much insulin as the Crisco rats to achieve this beautiful curve, nearly twice that in the VLC group (2.95ng/ml vs 1.79ng/ml, p<0.05).
So which rats are the most insulin sensitive? Not the Crisco rats. I'll accept that. Just say no to Crisco... It is completely arguable between the VCL and 60% carb group.
BUT. What would have happened if the VLC group had produced the same insulin response as the 60% carb group? Impossible, scream Axen and Axen. The VCL group have a blunted insulin response. It makes them well on the road to diabetes, metabolic syndrome, blindness, dialysis, we need the funding...
Calm down Peter, bit OTT there!
Except metabolic syndrome is characterised by elevated insulin, not depressed insulin. Duh.
I have to thank Helen who placed a comment on another post. She pointed out that glucokinase in the pancreas, the enzyme which the pancreas uses to sense glucose in the portal blood, is down regulated in response to carbohydrate restriction. Oh.
It is, err, up regulated in carbohydrate surplus.
This is what "bit" Axen and Axen in 2006. It looks like 15% of calories as carbohydrate in a VLC non-Crisco situation is adequate (on a high protein background) to allow pancreatic insulin secretion in response to glucose to become identical to that produced by rats on a 60% carbohydrate diet. Muscle TAG and associated molecules will drop too. Hence the overlay of the GTT curves in 2006.
Let us assume, very reasonably, that the VLC rats in 2010, on a 5% carb, energy restricted diet, are not expecting to deal with hyperglycaemia any time soon. They down regulate glucokinase production. Then some joker injects 1g/kg of glucose in to their peritoneal cavity. No one up regulates their glucokinase in 10 minutes, not even Super Rat*. Insulin response is blunted. Hyperglycaemia results.
*Actually Super Rat could do this but she is always busy saving the planet (again) and doesn't have time to help out here.
What would have happened with a few days carb loading in the VLC group before the GTT? Well, we (that "we" includes A&A) know the answer to this from 2006. Did you really think A&A are stupid? How many times do I have to point out that these people are good. Very, very good. They know that to get a "bad" result for VLC you must NOT increase carbs pre glucose load in a GTT.
Does anyone think that neither Axen nor Axen has heard of glucokinase? That would mean they're stupid. They're not, they know that if they allowed 15% carbs for a few days the VLC group would overlay the curve of the 60% carb group. For crying out loud, they published the damned curves themselves!
No. The effect of increased carbs on a VLC is not "unclear" (their word). It adjusts pancreatic insulin secretion to deal with carbs when carbs form a significant part of the diet. That's called physiology!
In summary:
Do A&A have a paradigm to support, a mortgage or two to pay, a living to make, careers to develop?
A few fatties getting injured is of no concern, provided the models can be adjusted to keep the funding coming through.
Will people edging towards type 2 diabetes get injured by a very low carbohydrate diet or will they be injured by A&A's funding success? What if they eat low fat high carbohydrate in the real world? What is hunger?
You decide. Then go eat some fat.
Peter
But not Crisco. Just say no....
Thursday, August 26, 2010
Here we go again

There might not be a lot of posts over the next few months. I'll get Axen and Axen (2) posted when I can!
Saturday, August 21, 2010
Axen and Axen (1)

Okay, the one without the chocolate all over their face is Ratty. He, Ratty, is 20 weeks old and weighs 360g. He is slim, active, well muscled (for a rat) and, of course, eats a very high fat diet. In fact he eats exactly the same food as we do with extra fried belly pork and cheese to snack on between meals of scrambled egg yolks (in butter), Bolognaise sauce, chilli mix or beef stew, should he ever get hungry between those main meals. My wife gave him a grape once. I removed it from his cage when it went mouldy.
I don't think he has metabolic syndrome. He eats an ad lib diet which provides about 70% of his calories from fat.
Now, I was looking through Axen and Axen's 2010 paper on managing metabolic syndrome in rats. First you have to make them obese. Hmmmm, now how might you do that? Ah ha! A high fat diet. In fact just 60% of calories will do the trick. Wow. Lets look at Table 1, column 2, HF diet used to induce obesity in rats. There it is: 60% fat, 25% protein and a mystery 15% carbohydrate.

Plus this line in the methods:
"The contents of saturated: monounsaturated: polyunsaturated fats were 25.3%:43.3%:31.3% in the HF diet and 31.7%:42.7%:25.5% in the VLC and HC diets (ω-3 fatty acids comprised ~2% of total fat)."
OK, are you thinking the carbohydrate was sucrose? Me too. Wrong! But the fat was supplied by Proctor and Gamble.
If you work through the paper to find out how this obesogenic diet was developed you get to ref 9, which is Axen and Axen again, this time in 2006. Once again, Table 1 for diet composition:

This tells us nothing more about the obesity diet (it's not really what the paper is about) but the fat was supplied by Proctor and Gamble. The paper does have another reference about the obesogenic diet, number 13 this time, which leads us to a gem from another Axen paper, just Kathleen and friends this time:
This is the sort of paper I love. Here is the abstract from 2003.
I think I have to put it here in full as it is really too important to summarise. They have a diet which develops metabolic syndrome in rats which CANNOT be prevented by restricting calories to normal body weight. Has any one heard of the "slim but metabolically obese" concept? The skinny type 2 diabetic? Kathleen Axen knows EXACTLY how to produce a skinny type 2 diabetic rat! And this is the paper. And here is the abstract:
"High fat, low carbohydrate diets are popularly advocated for weight loss and improvement in metabolic Syndrome X, a constellation of risk factors for type 2 diabetes mellitus and cardiovascular disease. The effects of an energy-restricted (to prevent weight gain in excess of normal growth) high fat (60% of energy), low carbohydrate (15%) diet were assessed in both lean rats and in rats previously rendered obese through ad libitum consumption of the same high fat diet. In obese rats, restriction of intake failed to improve impaired glucose tolerance, hyperinsulinemia, and hypertriglyceridemia, although it lowered visceral fat mass, liver lipid content and in vitro insulin hypersecretion compared with rats continuing to consume the high fat diet ad libitum. In lean rats, restricted intake of the high fat diet impaired glucose tolerance and increased visceral fat mass and liver lipid content. These findings support the conclusion that, in the absence of weight loss, a high fat, low carbohydrate diet not only may be ineffective in decreasing risk factors for cardiovascular disease and type 2 diabetes but may promote the development of disease in previously lower risk, nonobese individuals."
I think that that's pretty conclusive. A diet of 60% fat makes a rat glucose intolerant even if you starve it (and rats in the starved group will be HUNGRY, unlike Ratty) to a normal bodyweight. And no sucrose in sight. High fat and restricted carbohydrate diets might PROMOTE type two diabetes in rats (and people?) and give them heart attacks (do rats have heart attacks?) even if they were slim to begin with and stay slim on the diet. That's me and Ratty all right.
Ratty and I are doomed to diabetes it appears.
BTW, did I mention that Proctor and Gamble provided the fat?
Which fat? Well, here's the section which never made it in to the abstract:
"the other group was fed a high fat (HF;347 g fat/kg diet) low carbohydrate diet (22.6 kJ/g, 5.4 kcal/g). The HF diet was comprised of powdered Purina 5001 and hydrogenated vegetable fat (Proctor & Gamble, Cincinnati OH), with casein, L-methionine, AIN vitamin mix, and AIN mineral mix (Bio-serv, Frenchtown, NJ) (17) added to provide equivalent protein concentrations (LF, 234 g/kg diet; HF, 331 g/kg diet) and equivalent vitamin and mineral contents for the two diets. The hydrogenated vegetable fat contained 25% long-chain saturated, 44% monounsaturated and 28% PUFA, with 17% of total fat as trans fatty acids (manufacturer’s communication). This high fat, low carbohydrate diet was used because of the more pronounced obesity it has produced in rats in our laboratory than have several commercial high fat diets."
Stop, rewind, slow frame:
"with 17% of total fat as trans fatty acids"
That's a very special fat. Magic fat. Unless I am very much mistaken Proctor and Gamble make commercial bootpolish marketed as fit for human consumption. I think it's called Crisco. They have been poisoning America with it for decades.
DO NOT, UNDER ANY CIRCUMSTANCES, BASE YOUR DIET ON 60% OF CALORIES FROM FAT WHICH INCLUDES 17% TRANS FATTY ACIDS.
DO NOT DO IT.
Just say no.
But oh, oh, oh, Kathleen, why did you leave this out of the abstract????????????
People will think that eating fat makes you fat......
It certainly will if Proctor and Gamble supplied it.
Peter
OK, the 2006 and 2010 studies need looking at next in some detail. They are really quite funny in combination. Also, to give K Axen her due she does discuss trans fat toxicity extensively in the discussion of the paper. But not the abstract. There it's "fat" all the way. A pinguid diet (for Gary; ok do I win at Scrabble now?).
Actually, re reading the last sentence of the abstract: This is a very unpleasant piece of writing. Would people go as far as the full text to read what the paper is actually about?
Thoughts on problems with high fat diets
Back comments section of the uric acid post _Flo passed on the news of the death, due to stomach cancer, of a prominent long term Optimal Diet follower. This is not the sort of in formation that should be ignored although, from an individual case, I doubt whether there is anything we will every actually find out about causation. The main thing it brings home forcefully is that none of us is immortal and a good diet will not allow us to automatically reach an advanced age. The OD and similar approaches brings marked improvement in many of the diseases of civilisation but clearly not all. For someone who has achieved remission from multiple sclerosis or ankylosing spondylitis it is depressing to accept that those same diet changes might not be cancer protective, or not completely so.
_Flo has made her own modifications to her diet which are sensible in their own right and represent her choices. As regards the type of cancer it quite interesting to note that Poland has one of the highest rates of stomach cancer in the world. This snippet was passed along by another friend on the net and is corroborated by pubmed. It does suggest that speculation based on the location/type of the tumour may not be representative a specific diet related factor, rather the result of living in Poland. Of course it might be diet related.
I have my own personal tweaks to the OD and do not see any clear way of improving what I have already achieved.
So let be set here in print. No one has all of the answers.
I thought I would take this chance to discuss the possibility that there are people for whom low carbohydrate eating might be genuinely problematic. I have picked up a couple of hints along these lines over the years, so here we go with some thoughts.
I discussed a secondary prevention trial for heart disease here. All of these people had glucose regulation problems (that's part of my definition of having a heart attack). Adding 500-600kcal of either corn or olive oil to a very low fat diet precipitated diabetes in two of these individuals. Why? That's a good question. Type two diabetes is intrinsically linked to insulin resistance. Normally people eating a bolus of fat will reduce the rest of their calorie intake to compensate. Anyone who didn't would have placed a significant carbohydrate load on top of fat induced insulin resistance (even vegetable oils induce this, although palmitic acid does a rather better job) and failed to deal with their relatively high carbohydrate intake. Pure speculation but a low fat diet, with 80ml of added vegetable oil, undoubtedly triggered 2 cases of type two diabetes. I found that interesting.
Second hint was Jenny Ruhl's comment that she had come across very, very occasional individuals with diabetes who responded to low carbohydrate eating by deterioration of glycaemic control. Again, I have no details what so ever but anyone's ears should prick up when they hear things like this. Denial is not where it's at.
Then Carb Sane introduced me to the Otsuka Long-Evans Tokushima Fatty strain (OLETF) of rats. These rats are a diabetologists dream. Under fixed isocaloric conditions they gain weight and fat mass in direct proportion to the percentage of fat in an exactly measured 28.7 joule daily ration. Like, wow. Fat really does make you fat!
Of course the OLETF rat is described as a Good Model for human type two diabetes. It explains exactly why all diabetics put on to a LC, high fat diet become obese and hyperglycaemiac. What do you mean, they don't? Oh, not enough testosterone! The OLETF rats only become diabetic if they are male. That's why all type two diabetics are blokes. What do you mean, women get type two diabetes? But out model says only blokes should. And it's a Good Model...
OK facetiousness aside, what is happening in the OLETF rats? Is it possible that there are some humans out there with OLEFT rat style type two diabetes. Well, why not?
Let's have a look at what is happening from the point of view that insulin is the primary hormone in the development of obesity. If you think about someone eating just once a day, essentially all of their calories are going to get stored. Glycogen in the liver and fat in the fat. What determines weight gain is how much of that stored energy fails to be extracted from storage before the next meal arrives.
Summary: No one stores lipids or glucose in their blood stream. It all goes in to short term storage. What comes out determines weight loss. Insulin determines what comes out.
OLETF rats, on high fat, fixed calorie diet of 28.7 joules per day put all of this energy in to storage but fail to extract as many of these calories/joules from storage as those OLETF rats on 28.7 joules of a high starch diet. What is going on?
If you accept the insulin hypothesis of weight gain, the answer is that dietary fat is being trapped in adipocytes by excessive blood insulin. There must be excess insulin.
Why is the insulin elevated when there is a reduced dietary stimulus for insulin production? These rats have peripheral, almost certainly muscle based, insulin resistance. But only on a high fat diet.
High fat diets put lipids in to muscles, muscles full of lipid don't accept glucose. If the system works correctly the muscles run on lipids until there is a balance between fat supply rejecting glucose and fat depletion allowing glucose acceptance. A few billion years is ample time to get this system working correctly to maintain normoglycaemia in the face of varied macronutrient intakes from day to day.
What might be broken in this system in the OLETF rat?
Well, if you can get lipids in to muscle cells but cannot then use that lipid for beta oxidation you would expect to develop muscle insulin resistance in proportion to the amount of fat you supply, ie if the fat enters the muscles but does not go any further those muscles will become insulin resistant and stay insulin resistant. If muscles are not accepting glucose because they are insulin resistant the glucose is going to have to be dealt with by increased levels of insulin. Hyperglycaemia is unacceptable.
The extra insulin needed to maintain normoglycaemia then traps stored dietary fat in adipocytes. The rats get fat because they cannot get fat out of adipocytes. They will also do less running around and will probably feel colder than normal rats because they have no access to their fatty tissue for energy supplies. If they had access to food they would eat more, but 28.7 joules per day was the limit in this experiment. If they had access to more calories they would clearly eat more because no one likes the hunger and shivering produced by sequestering a chunk of your caloric fat intake in your adipocytes and locking it in there with insulin.
Once your fat cells get full enough they will spill free fatty acids because they are now too full to listen to insulin any more. These FFAs join those intra cellular muscle tissue di and tri glycerides from the metabolic defect. Intra myocyte fatty acids still have no where to go, so muscle insulin resistance rockets, plasma glucose rockets and you have a superb model of fat induced obesity and peripheral insulin resistance.
Glucose enters mitochondria as pyruvate. Fatty acids enter mitochondria as acyl CoA moieties. The place to be looking for explanations for the syndrome seen in the OLETF rat is in fatty acid processing. My guess is that lipid molecules get in to myocytes but never get effectively passed to the mitochondria. Quite what testosterone has to do with this is beyond me, it's not me that is suggesting the OLETF rat is a good model for human type 2 diabetes!
You have to ask what would have happened on a ketogenic diet. Would ketosis have side stepped this problem? Ketone bodies enter mitochondria without any need for long chain fatty acid transporters. They use monocarboxylate transporters, just like pyruvate... You don't really think that Kaneko et al would so something as stupid as putting an OLETF rat on a ketogenic diet? But it would have been interesting. I'm not sure it would side step the problem but it might. I'm certainly not expecting a diabetologist to find out for me.
I really enjoyed the OLETF rat. Does it tell me something about type two diabetes? Only that there might be very, very special people who respond to dietary fat with hyperglycaemia.
I never did find any teeth in my chickens. I understand hen's teeth are rare. So are OLETF humans.
But they probably exist (not the hen's teeth!).
Peter
_Flo has made her own modifications to her diet which are sensible in their own right and represent her choices. As regards the type of cancer it quite interesting to note that Poland has one of the highest rates of stomach cancer in the world. This snippet was passed along by another friend on the net and is corroborated by pubmed. It does suggest that speculation based on the location/type of the tumour may not be representative a specific diet related factor, rather the result of living in Poland. Of course it might be diet related.
I have my own personal tweaks to the OD and do not see any clear way of improving what I have already achieved.
So let be set here in print. No one has all of the answers.
I thought I would take this chance to discuss the possibility that there are people for whom low carbohydrate eating might be genuinely problematic. I have picked up a couple of hints along these lines over the years, so here we go with some thoughts.
I discussed a secondary prevention trial for heart disease here. All of these people had glucose regulation problems (that's part of my definition of having a heart attack). Adding 500-600kcal of either corn or olive oil to a very low fat diet precipitated diabetes in two of these individuals. Why? That's a good question. Type two diabetes is intrinsically linked to insulin resistance. Normally people eating a bolus of fat will reduce the rest of their calorie intake to compensate. Anyone who didn't would have placed a significant carbohydrate load on top of fat induced insulin resistance (even vegetable oils induce this, although palmitic acid does a rather better job) and failed to deal with their relatively high carbohydrate intake. Pure speculation but a low fat diet, with 80ml of added vegetable oil, undoubtedly triggered 2 cases of type two diabetes. I found that interesting.
Second hint was Jenny Ruhl's comment that she had come across very, very occasional individuals with diabetes who responded to low carbohydrate eating by deterioration of glycaemic control. Again, I have no details what so ever but anyone's ears should prick up when they hear things like this. Denial is not where it's at.
Then Carb Sane introduced me to the Otsuka Long-Evans Tokushima Fatty strain (OLETF) of rats. These rats are a diabetologists dream. Under fixed isocaloric conditions they gain weight and fat mass in direct proportion to the percentage of fat in an exactly measured 28.7 joule daily ration. Like, wow. Fat really does make you fat!
Of course the OLETF rat is described as a Good Model for human type two diabetes. It explains exactly why all diabetics put on to a LC, high fat diet become obese and hyperglycaemiac. What do you mean, they don't? Oh, not enough testosterone! The OLETF rats only become diabetic if they are male. That's why all type two diabetics are blokes. What do you mean, women get type two diabetes? But out model says only blokes should. And it's a Good Model...
OK facetiousness aside, what is happening in the OLETF rats? Is it possible that there are some humans out there with OLEFT rat style type two diabetes. Well, why not?
Let's have a look at what is happening from the point of view that insulin is the primary hormone in the development of obesity. If you think about someone eating just once a day, essentially all of their calories are going to get stored. Glycogen in the liver and fat in the fat. What determines weight gain is how much of that stored energy fails to be extracted from storage before the next meal arrives.
Summary: No one stores lipids or glucose in their blood stream. It all goes in to short term storage. What comes out determines weight loss. Insulin determines what comes out.
OLETF rats, on high fat, fixed calorie diet of 28.7 joules per day put all of this energy in to storage but fail to extract as many of these calories/joules from storage as those OLETF rats on 28.7 joules of a high starch diet. What is going on?
If you accept the insulin hypothesis of weight gain, the answer is that dietary fat is being trapped in adipocytes by excessive blood insulin. There must be excess insulin.
Why is the insulin elevated when there is a reduced dietary stimulus for insulin production? These rats have peripheral, almost certainly muscle based, insulin resistance. But only on a high fat diet.
High fat diets put lipids in to muscles, muscles full of lipid don't accept glucose. If the system works correctly the muscles run on lipids until there is a balance between fat supply rejecting glucose and fat depletion allowing glucose acceptance. A few billion years is ample time to get this system working correctly to maintain normoglycaemia in the face of varied macronutrient intakes from day to day.
What might be broken in this system in the OLETF rat?
Well, if you can get lipids in to muscle cells but cannot then use that lipid for beta oxidation you would expect to develop muscle insulin resistance in proportion to the amount of fat you supply, ie if the fat enters the muscles but does not go any further those muscles will become insulin resistant and stay insulin resistant. If muscles are not accepting glucose because they are insulin resistant the glucose is going to have to be dealt with by increased levels of insulin. Hyperglycaemia is unacceptable.
The extra insulin needed to maintain normoglycaemia then traps stored dietary fat in adipocytes. The rats get fat because they cannot get fat out of adipocytes. They will also do less running around and will probably feel colder than normal rats because they have no access to their fatty tissue for energy supplies. If they had access to food they would eat more, but 28.7 joules per day was the limit in this experiment. If they had access to more calories they would clearly eat more because no one likes the hunger and shivering produced by sequestering a chunk of your caloric fat intake in your adipocytes and locking it in there with insulin.
Once your fat cells get full enough they will spill free fatty acids because they are now too full to listen to insulin any more. These FFAs join those intra cellular muscle tissue di and tri glycerides from the metabolic defect. Intra myocyte fatty acids still have no where to go, so muscle insulin resistance rockets, plasma glucose rockets and you have a superb model of fat induced obesity and peripheral insulin resistance.
Glucose enters mitochondria as pyruvate. Fatty acids enter mitochondria as acyl CoA moieties. The place to be looking for explanations for the syndrome seen in the OLETF rat is in fatty acid processing. My guess is that lipid molecules get in to myocytes but never get effectively passed to the mitochondria. Quite what testosterone has to do with this is beyond me, it's not me that is suggesting the OLETF rat is a good model for human type 2 diabetes!
You have to ask what would have happened on a ketogenic diet. Would ketosis have side stepped this problem? Ketone bodies enter mitochondria without any need for long chain fatty acid transporters. They use monocarboxylate transporters, just like pyruvate... You don't really think that Kaneko et al would so something as stupid as putting an OLETF rat on a ketogenic diet? But it would have been interesting. I'm not sure it would side step the problem but it might. I'm certainly not expecting a diabetologist to find out for me.
I really enjoyed the OLETF rat. Does it tell me something about type two diabetes? Only that there might be very, very special people who respond to dietary fat with hyperglycaemia.
I never did find any teeth in my chickens. I understand hen's teeth are rare. So are OLETF humans.
But they probably exist (not the hen's teeth!).
Peter
Friday, August 06, 2010
Urate, ascorbate, resveratrol and land mines
It only needs a brief glance through the literature to realise that uric acid is a prime mover in metabolic syndrome and might reasonably compete with cholesterol as the premier mammalian-synthesised molecule of self destruction.
Until of course you come across interesting papers like this one which suggests that uric acid is a signal of tissue injury, a mobiliser of repair systems and, in particular, a recruiter of endothelial progenitor cells.
In modern terms, stepping on a land-mine will produce a surge of uric acid to blunt the effects of the renal ischaemia which will occur as you bleed out through the remains of where your foot used to be. In evolutionary terms you can see how surviving acute trauma might be beneficial and it looks like uric acid is seriously useful in this context. Stepping on a land mine every day fairly rapidly becomes problematic in terms of all cause mortality and even uric acid is unlikely to maintain its efficacy with prolonged usage. Chronic drug induced hyper uricaemia appears to be a Bad Thing in that it blunts the benefits of acute elevations.

If you don't live in a heavily land-mined country you are (a) lucky and (b) more likely to be injured by a bottle of cola or a bowl of apples. Or maybe oranges, stawberries, kiwis etc. Whether it is simply the fructose in the cola or some other plant nasty in the fruit in addition to fructose isn't particularly clear. But your body produces a spike of uric acid in response. It's been injured.
It is perfectly possible to consume cola or fruit on a chronic basis without the immediately obvious effects of repeatedly treading on a land mine.
This leads me to whether there are direct benefits from minor damage of eating fruit via hormesis or whether it is just all bad. I suspect it depends on the dose and the chronicity. When you look at WHEL and PPT there may well be some sort of accommodation which leaves total mortality unchanged by eight years of eating extra fruit and veg. There seems to be little doubt that an acute rise in uric acid is beneficial and chronically elevated uric acid may be less so. This flies in the face of Kwasniewski's opinions about uric acid, the more the better. But he is talking about elevated uric acid in a LC high saturated fat situation, not after eating a bowl of apples a day for a few years.... Of course he never cites references but I'm keeping an eye out!
Ascorbate is interesting as it is a rather ubiquitous antioxidant and does appear to be used by humans for assorted essential functions, all be it in very small amounts, despite the fact we cannot synthesise it. Most mammals tightly regulate their ascorbate production to their needs as they can produce it on demand. Not so humans. We don't produce it and our intake is completely random, varying from 10mg per day or less on an all meat diet to a few 1000mgs if you fall face down in a clump of cranberries and keep eating all day.
The chances of you meeting the cranberries every day for six weeks are slim, so if you want to study the effect of relatively high doses of ascorbate on free radical mediated processes in humans you have to use supplements. Eating 1000mg represents an awful lot of berries but is a common supplement dose.
This group in Spain actually pre empted the group in Germany in demonstrating the adverse effects of ascorbate supplementation on the effects of exercise.
Exercise produces free radicals, free radicals signal for both muscle cell division and mitochondrial number increase (using the surrogate of cytochrome C for mitochondrial number).

If you dose with 1000mg ascorbate daily you will scavenge the free radicals and blunt the increase in both muscle mass and mitochondrial number which should have been generated by those free radicals. You might expect this and it's worth noting that the effect appears to clearly detectable but far from complete.
I have worked on the assumption that taking antioxidants should down regulate your own endogenous antioxidant production. The Spanish group used rats for this part of their research and did indeed find that gene expression for synthesis of both superoxide dismutase and glutathione peroxidase are markedly down regulated. A zero increase with ascorbate supplemented exercise compared to a 3.5 fold increase after three weeks of unsupplemented exercise.

Think of the implications. You exercise, exercise generates free radicals, free radicals build muscle and upregulate production of antioxidant enzymes. So exercise is, fundamentally, antioxidant (while ever it is kept within the limits which can be accommodated by synthesis of SOD and GPx). Obviously ultra-marathon runners must be willing to injure themselves for their sport.
Mega dosing with ascorbate markedly blunts the induction of the intrinsic antioxidant system of mammals, even of a rat which naturally produces ascorbate.
What about resveratrol? Feed it to a mouse in quantities equivalent to between 150 and 1,500 bottles of wine a day and you get some improvement in inflammatory markers. Do you simultaneously down regulate SOD and GPx synthesis? Now there is a question!
That will be a cracking study. The answer will be yes if resveratrol really scavenges significant numbers free radicals. Our own endogenous antioxidant systems are fine tuned to our needs. What does a blanket treatment with a highly potent antioxidant do to our ability to respond to the daily alterations in free radicals? Free radicals carry information. How do we replace the information lost if they are stuck to a resveratrol molecule?
Luckily our bodies try hard not to absorb the stuff and normally metabolise it to sulphated or glucuronated forms within minutes. It's unlikely that we ever have much free resveratrol (or dark chocolate favanoids either, phew) in our bloodstream until human ingenuity invented the resveratrol chewing gum.
Unintended consequences anyone?
Peter
BTW The Wiki article on resveratrol appears to be very contentious. Wiki becomes a battleground once you get on to sticky subjects like cholesterol and, apparently, resveratrol. I've kept the text for when it gets edited to "Resveratrol explains the French paradox and will make us live for ever"
Until of course you come across interesting papers like this one which suggests that uric acid is a signal of tissue injury, a mobiliser of repair systems and, in particular, a recruiter of endothelial progenitor cells.
In modern terms, stepping on a land-mine will produce a surge of uric acid to blunt the effects of the renal ischaemia which will occur as you bleed out through the remains of where your foot used to be. In evolutionary terms you can see how surviving acute trauma might be beneficial and it looks like uric acid is seriously useful in this context. Stepping on a land mine every day fairly rapidly becomes problematic in terms of all cause mortality and even uric acid is unlikely to maintain its efficacy with prolonged usage. Chronic drug induced hyper uricaemia appears to be a Bad Thing in that it blunts the benefits of acute elevations.

If you don't live in a heavily land-mined country you are (a) lucky and (b) more likely to be injured by a bottle of cola or a bowl of apples. Or maybe oranges, stawberries, kiwis etc. Whether it is simply the fructose in the cola or some other plant nasty in the fruit in addition to fructose isn't particularly clear. But your body produces a spike of uric acid in response. It's been injured.
It is perfectly possible to consume cola or fruit on a chronic basis without the immediately obvious effects of repeatedly treading on a land mine.
This leads me to whether there are direct benefits from minor damage of eating fruit via hormesis or whether it is just all bad. I suspect it depends on the dose and the chronicity. When you look at WHEL and PPT there may well be some sort of accommodation which leaves total mortality unchanged by eight years of eating extra fruit and veg. There seems to be little doubt that an acute rise in uric acid is beneficial and chronically elevated uric acid may be less so. This flies in the face of Kwasniewski's opinions about uric acid, the more the better. But he is talking about elevated uric acid in a LC high saturated fat situation, not after eating a bowl of apples a day for a few years.... Of course he never cites references but I'm keeping an eye out!
Ascorbate is interesting as it is a rather ubiquitous antioxidant and does appear to be used by humans for assorted essential functions, all be it in very small amounts, despite the fact we cannot synthesise it. Most mammals tightly regulate their ascorbate production to their needs as they can produce it on demand. Not so humans. We don't produce it and our intake is completely random, varying from 10mg per day or less on an all meat diet to a few 1000mgs if you fall face down in a clump of cranberries and keep eating all day.
The chances of you meeting the cranberries every day for six weeks are slim, so if you want to study the effect of relatively high doses of ascorbate on free radical mediated processes in humans you have to use supplements. Eating 1000mg represents an awful lot of berries but is a common supplement dose.
This group in Spain actually pre empted the group in Germany in demonstrating the adverse effects of ascorbate supplementation on the effects of exercise.
Exercise produces free radicals, free radicals signal for both muscle cell division and mitochondrial number increase (using the surrogate of cytochrome C for mitochondrial number).

If you dose with 1000mg ascorbate daily you will scavenge the free radicals and blunt the increase in both muscle mass and mitochondrial number which should have been generated by those free radicals. You might expect this and it's worth noting that the effect appears to clearly detectable but far from complete.
I have worked on the assumption that taking antioxidants should down regulate your own endogenous antioxidant production. The Spanish group used rats for this part of their research and did indeed find that gene expression for synthesis of both superoxide dismutase and glutathione peroxidase are markedly down regulated. A zero increase with ascorbate supplemented exercise compared to a 3.5 fold increase after three weeks of unsupplemented exercise.

Think of the implications. You exercise, exercise generates free radicals, free radicals build muscle and upregulate production of antioxidant enzymes. So exercise is, fundamentally, antioxidant (while ever it is kept within the limits which can be accommodated by synthesis of SOD and GPx). Obviously ultra-marathon runners must be willing to injure themselves for their sport.
Mega dosing with ascorbate markedly blunts the induction of the intrinsic antioxidant system of mammals, even of a rat which naturally produces ascorbate.
What about resveratrol? Feed it to a mouse in quantities equivalent to between 150 and 1,500 bottles of wine a day and you get some improvement in inflammatory markers. Do you simultaneously down regulate SOD and GPx synthesis? Now there is a question!
That will be a cracking study. The answer will be yes if resveratrol really scavenges significant numbers free radicals. Our own endogenous antioxidant systems are fine tuned to our needs. What does a blanket treatment with a highly potent antioxidant do to our ability to respond to the daily alterations in free radicals? Free radicals carry information. How do we replace the information lost if they are stuck to a resveratrol molecule?
Luckily our bodies try hard not to absorb the stuff and normally metabolise it to sulphated or glucuronated forms within minutes. It's unlikely that we ever have much free resveratrol (or dark chocolate favanoids either, phew) in our bloodstream until human ingenuity invented the resveratrol chewing gum.
Unintended consequences anyone?
Peter
BTW The Wiki article on resveratrol appears to be very contentious. Wiki becomes a battleground once you get on to sticky subjects like cholesterol and, apparently, resveratrol. I've kept the text for when it gets edited to "Resveratrol explains the French paradox and will make us live for ever"
Sunday, August 01, 2010
What's the secret of time travel doing on Fry's ass?
I would just like to paraphrase that relevant line from Futurama:
What's the secret of Eternal Youth doing on a grapeskin? It was bound to be somewhere..........
Resveratrol. Gift of plants. Oh oh, wrong again!
Many thanks to Blogblog for the link to that hysterical abstract, I just love it. If anyone thinks that resveratrol is the answer to the French Paradox, or that there even is a French Paradox, or that plants will gift us eternal youth, then you're in the wrong place on the net I'm afraid, just hit the back button!
Peter
My penance for cribbing 7.9 seconds from the Futurama movie: "Buy it, buyyyyyyy it......."
Wednesday, July 28, 2010
Fruit and vegetables (12) WHEL and CVD
I just thought I'd have a quick look (after viewing the PPT trawls) at the data trawls from WHEL in case they'd looked at the effect of a diet high in fruit 'n' fibre and low in fat on CVD. Of course they did. It gets kind of boring trotting out these bias confirming studies, but here's the punchline anyway:
"Over a mean of 8.1 years, a dietary intervention that reduced total fat intake and increased intakes of vegetables, fruits, and grains did not significantly reduce the risk of CHD, stroke, or CVD in postmenopausal women..."
Maybe they need more cream on the fruit? I'm just so grateful that they didn't get anything correct by accident. WHEL and PPT are such great sorces of hard data on crap diets.
Peter
"Over a mean of 8.1 years, a dietary intervention that reduced total fat intake and increased intakes of vegetables, fruits, and grains did not significantly reduce the risk of CHD, stroke, or CVD in postmenopausal women..."
Maybe they need more cream on the fruit? I'm just so grateful that they didn't get anything correct by accident. WHEL and PPT are such great sorces of hard data on crap diets.
Peter
Monday, July 26, 2010
Jimmy Moore on Dr Weil
Another one liner in case you don't follow Jimmy Moore, the post on Dr Weil is very interesting from the political point of view. Very interesting.
Peter
Peter
Sunday, July 25, 2010
Wheat and lactase
While I'm trying to clear my desk top a little, with limited access to the net during an oncall weekend, I thought I might post this link as I'm thinking wheat at the moment.
I think I've discussed the role of gluten in trashing the brush border cells, again without need for any sort of immune system involvement. The present study did involve immune-injured people but I see no reason why exactly the same principle shouldn't apply to antibody negative "coeliacs" (suffering from "imagination" or even, gasp, orthorexia nervosa, another from Elizabeth. Apologies for the picture of the bowl of rabbit food).
Another aspect of normalising the brush border is that far less food is going to be left in the small intestine to feed a bacterial overgrowth. On a LC, low fibre diet there are likely to be massive changes in gut flora. I have an anecdote from several years ago given to me by a lady and her mother who had gone LC plus gluten free and, after quite some time, both suddenly became lactose intolerant. This is the last thing I would have expected.
Now, I'm no great enthusiast for pre- or pro-biotics, but I do eat a fair amount of cheese, soured cream and high fat yogurt. The last of those occasionally in large amounts. So eating real-food dairy accidentally includes plenty of germs. Germs to which we are well adapted. Gut bacteria are normal to humans and lactobacilli are amongst the first bacteria to colonise the gut of a healthy infant, presumably because its mother provides lactose (and pre-biotics too, hmmmm). For humans who are not genetically adapted to adult lactose intake, are we borrowing the gene for lactose tolerance from the microbiome in our gut? Is it possible to lose lactose tolerance if we lose lactobacilli during the major bacterial starve-out that LC probably produces?
Alternatively, as coeliac disease appear to require certain bacteria, does lactose intolerance REQUIRE certain non-lactobacilli bacteria? This would fit better with two people developing lactose intolerance at the same time in the same household, if they simultaneously picked up the same bacterial opportunist.
Answers on a postcard to........
Peter
I think I've discussed the role of gluten in trashing the brush border cells, again without need for any sort of immune system involvement. The present study did involve immune-injured people but I see no reason why exactly the same principle shouldn't apply to antibody negative "coeliacs" (suffering from "imagination" or even, gasp, orthorexia nervosa, another from Elizabeth. Apologies for the picture of the bowl of rabbit food).
Another aspect of normalising the brush border is that far less food is going to be left in the small intestine to feed a bacterial overgrowth. On a LC, low fibre diet there are likely to be massive changes in gut flora. I have an anecdote from several years ago given to me by a lady and her mother who had gone LC plus gluten free and, after quite some time, both suddenly became lactose intolerant. This is the last thing I would have expected.
Now, I'm no great enthusiast for pre- or pro-biotics, but I do eat a fair amount of cheese, soured cream and high fat yogurt. The last of those occasionally in large amounts. So eating real-food dairy accidentally includes plenty of germs. Germs to which we are well adapted. Gut bacteria are normal to humans and lactobacilli are amongst the first bacteria to colonise the gut of a healthy infant, presumably because its mother provides lactose (and pre-biotics too, hmmmm). For humans who are not genetically adapted to adult lactose intake, are we borrowing the gene for lactose tolerance from the microbiome in our gut? Is it possible to lose lactose tolerance if we lose lactobacilli during the major bacterial starve-out that LC probably produces?
Alternatively, as coeliac disease appear to require certain bacteria, does lactose intolerance REQUIRE certain non-lactobacilli bacteria? This would fit better with two people developing lactose intolerance at the same time in the same household, if they simultaneously picked up the same bacterial opportunist.
Answers on a postcard to........
Peter
Wheat Germ Agglutinin; how little is enough?
Chignola's group have been at WGA again. Their basic interest is (was?) in using WGA as a drug carrier aimed at certain cell surface sugar groupings, hoping to target certain cancers, as far as I can see.
Their first paper in 2005 demonstrated direct intestinal cell toxicity at micro molar concentrations. However Glenn updated me on their more recent 2009 paper here:
"At nanomolar concentrations WGA stimulates the synthesis of pro-inflammatory cytokines and thus the biological activity of WGA should be reconsidered by taking into account the effects of WGA on the immune system at the gastrointestinal interface. These results shed new light onto the molecular mechanisms underlying the onset of gastrointestinal disorders observed in vivo upon dietary intake of wheat-based foods"
Notice there is no mention of coeliac disease, you do not need to be genetically predisposed by HLA type to have WGA toxicity. This looks to be yet more direct molecular toxicity.
Now we are talking nanomolar concentrations.
It's a bit difficult to get your head round a nanomole of WGA. One mole is the molecular weight expressed in grams. For WGA this is a BIG number of grams. A nanomole is not very much. So a nanomole is not very much of quite a lot of grams! Without struggling with the math I'd just suggest putting the bread in the bin and seeing if I could get out of my wheelchair as my gut integrity improved! An angry immune cell, looking at a soup of gut peptides, is not going to care what collateral damage it does.
Peter
BTW I heard that Sheffield vs Nottingham medics is generic and not limited to neurology... I can't remember who mentioned it, I'm pretty certain it was off blog at a supper party, from a physicist who had worked for Nottingham Queens Medical in some capacity.
Their first paper in 2005 demonstrated direct intestinal cell toxicity at micro molar concentrations. However Glenn updated me on their more recent 2009 paper here:
"At nanomolar concentrations WGA stimulates the synthesis of pro-inflammatory cytokines and thus the biological activity of WGA should be reconsidered by taking into account the effects of WGA on the immune system at the gastrointestinal interface. These results shed new light onto the molecular mechanisms underlying the onset of gastrointestinal disorders observed in vivo upon dietary intake of wheat-based foods"
Notice there is no mention of coeliac disease, you do not need to be genetically predisposed by HLA type to have WGA toxicity. This looks to be yet more direct molecular toxicity.
Now we are talking nanomolar concentrations.
It's a bit difficult to get your head round a nanomole of WGA. One mole is the molecular weight expressed in grams. For WGA this is a BIG number of grams. A nanomole is not very much. So a nanomole is not very much of quite a lot of grams! Without struggling with the math I'd just suggest putting the bread in the bin and seeing if I could get out of my wheelchair as my gut integrity improved! An angry immune cell, looking at a soup of gut peptides, is not going to care what collateral damage it does.
Peter
BTW I heard that Sheffield vs Nottingham medics is generic and not limited to neurology... I can't remember who mentioned it, I'm pretty certain it was off blog at a supper party, from a physicist who had worked for Nottingham Queens Medical in some capacity.
Thursday, July 22, 2010
Total Perspective Vortex and vegicide
For those who have failed to read the Hitchhiker's Guide to the Galaxy a little information about the Total Perspective Vortex is required. It's in context here.
The relevant quote is this one:
"To Trin Tragula’s horror, the shock completely annihilated her [his wife's] brain; but to his satisfaction he realized that he had proved conclusively that if life is going to exist in a Universe of this size, then the one thing it cannot have is a sense of proportion"
I hate to mention anyone with an annihilated brain after mentioning poor Dr Stanton, but here is the key quote from Dr T Colon Campbell (none of the rest is worth reading):
"In summary, Denise’s critique lacks a sense of proportionality."
Phew, just as well, we need Denise with a non annihilated brain to crunch numbers for us.
But the implication, unstated, is that Dr Colon has a sense of proportionality.
Explains a lot really.
Peter
BTW Zaphod eats the cake. There, ruined the series for you!
The relevant quote is this one:
"To Trin Tragula’s horror, the shock completely annihilated her [his wife's] brain; but to his satisfaction he realized that he had proved conclusively that if life is going to exist in a Universe of this size, then the one thing it cannot have is a sense of proportion"
I hate to mention anyone with an annihilated brain after mentioning poor Dr Stanton, but here is the key quote from Dr T Colon Campbell (none of the rest is worth reading):
"In summary, Denise’s critique lacks a sense of proportionality."
Phew, just as well, we need Denise with a non annihilated brain to crunch numbers for us.
But the implication, unstated, is that Dr Colon has a sense of proportionality.
Explains a lot really.
Peter
BTW Zaphod eats the cake. There, ruined the series for you!
Tuesday, July 20, 2010
Where is the brain of Rosemary Stanton?
I've posted on Dr Peter Clifton in the past. He's in the news again. Here's the link to the Telegraph article from Blogblog plus the text is also over on the trialblog archive for when the link goes down. Obviously there is a limit to the functionality of his brain implant (inability to perceive that obesity is a symptom not a disease for example, still pro vegetables because they are low in calories, no idea of the role of insulin in obesity etc) but he does seem to be able to conceive that five a day of fruit 'n' veg are a waste of money. That is utterly amazing.
Hmmmmm. On re reading the article, apart from finding fruit and veg useless, he still seems utterly lost as far as meaningful comprehension goes.
Still, Clifton's brain implant has some residual functionality. But where did the brain come from???? Could it have been stolen from Dr Rosemary Stanton? The Dr as in the last sentence in the Telegraph article:
"Nutritionist Dr Rosemary Stanton cast doubt on the findings and suggested the study [Clifton's] could be flawed."
Her brain could easily have been taken as the source of whatever residual central processing power is available to Dr Clifton. Replacing Dr Stanton's brain with a low IQ parrot would leave her fully functional as a nutritionist... It all fits together.
Peter
Hmmmmm. On re reading the article, apart from finding fruit and veg useless, he still seems utterly lost as far as meaningful comprehension goes.
Still, Clifton's brain implant has some residual functionality. But where did the brain come from???? Could it have been stolen from Dr Rosemary Stanton? The Dr as in the last sentence in the Telegraph article:
"Nutritionist Dr Rosemary Stanton cast doubt on the findings and suggested the study [Clifton's] could be flawed."
Her brain could easily have been taken as the source of whatever residual central processing power is available to Dr Clifton. Replacing Dr Stanton's brain with a low IQ parrot would leave her fully functional as a nutritionist... It all fits together.
Peter
Friday, July 16, 2010

Thursday, July 15, 2010
Polyp Prevention Trial and homocysteine
I don't know where to start with this. Let's begin with one of my all time favourite studies, the Polyp Prevention Trial, PPT. For those of us who have forgotten, this quote from the abstract sums up PPT:
"This study failed to show any effect of a low-fat, high-fiber, high-fruit and -vegetable eating pattern on adenoma recurrence even with 8 years of follow-up"
Now, would anyone like to guess how much the PPT cost? I've no idea, but I guess it cost enough that you are not going to accept a result like that after spending all of those tax dollars (thank goodness it wasn't pounds sterling, it's bad enough funding the FSA on this side of the Pond). So there is now an on-going avalanche of subgroup analysis papers flooding out from PPT, many of which are utterly hysterical. Some actually show, by accident, the injury done to many of the people in the intervention arm of the study. From these new papers you would NEVER guess the main trial primary end point was an utter destruction of the hypothesis that flatulence is good for you.
Let's start with that lovely sub group who ate all the dry beans. Upping your beans by 370% appeared to drop your risk for adenoma recurrence to an OR of 0.35 in the intervention arm. This is music to the fibre-ophile ear. The effect is massive. It really should show in observational studies if it is real. It doesn't.
Sub group analysis of the control group, observational only, suggests that contributing to global warming is NOT justified for colorectal cancer prevention. There is no excuse for flatulence. I dunno. Maybe the intervention trial is correct. But don't hold your breath, just your nose.
That last sub group analysis is particularly interesting once you get hold of the full text (many thanks Anna). It only looks at the control arm, so it's observational in nature. They blood sampled folks at the start of the study then counted polyps after four years on whatever shade of the SAD happened to be chosen by people who have had a polyp removed but no diet advice given (ie the lucky ones).
What is topical to me at the moment is that homocysteine (HCy) correlation with polyp recurrence. HCy at enrolment is a very good predictor of polyp recurrence in this group of untutored polyp formers over the next 4 years.
The suggestion, by the authors, is that reducing HCy will reduce adenoma recurrence. Really, someone should test this hypothesis. Maybe a trial increasing fruit, vegetables and fibre should be done to see if these miracle foods will reduce adenoma formation over a four or even eight year period?
Oh, it's been done has it? Ah yes, that's where this data trawl has come from! I remember, yes, that's it, eight years of fruit and fibre does bugger all to prevent polyp recurrence. Silly of me to forget. There was an intervention arm to PPT!
Remember that and re read the very carefully worded abstract... Would you have guessed?
Before I get on to a dietary intervention which might lower HCy (if you care, all this is observational), let's have a look at another subgroup analysis of the PPT data.
I now want to look at what happened in the intervention arm. We can tease out a sub group, the folks who really complied with the intervention, and compare them with the people in the intervention arm who complied poorly.
That's this subgroup analysis.
A total of 210 "super compliers" were noted and compared to controls. I can't really tell if the controls were on the SAD or were those less compliant members of the intervention arm, probably the latter. The odds ratio for adenoma recurrence of supercompliance was 0.65 and statistically significant.
So 210 people out of the 1,905 in the intervention arm benefited. That means that 1,695 people didn't benefit from four years of advice.
But hold on a minute. When the initial study compared intervention with non intervention there was no benefit from intervention compared to the SAD. So if 210 people did better than average, then 1,695 people MUST HAVE DONE WORSE than average. That is, they did worse than those on the SAD. You would have expected a 50:50 split, obviously half should do better and half should do worse than average!
EDIT: Of course if they compared 210 super compliers to the 1,695 injured people they possibly got p < 0.05 because these 1,695 did particularly badly. I wonder if p would have been < 0.05 if they had compared the 210 to the whole SAD group? Probably not would be my guess. Or they would have done it! Me, cynical? Surely not... END EDIT
We can summarise: 1,695 people were injured by being in the intervention arm of the PPT.
I would also point out that "super compliers" are super compliers. These are the sort of people who meticulously take their placebo tablets in clinical trials and consistently do better than those who are lackadaisical with consuming their placebo tablets. Mike Eades has blogged about it nicely here.
So did the 210 super compliers benefit from the diet or were they just the sort of people who comply with authority and automatically do well despite their doctor's best efforts? There was no scope for an adherer effect in the SAD non-intervention arm because they were left alone with no religious group therapy support. Oops, I mean dietary advice...
So was everyone injured in the intervention group but a few had their injury offset by the warm glow of study compliance?
Don't hold your breath waiting to see THAT get discussed in an NIH funded paper. We're looking to justify the cash spent on a superb study which has made no friends among people with control over the purse strings...
Anyhow, back to HCy. We all know from nauseous observational stuff that PPT intervention should have dropped HCy (I've not seen this analysis anywhere yet but I guess it did) but it still didn't drop adenoma recurrence and it probably worsened it in that vast majority of the participants who weren't super compliers.
What sort of HCy levels are present in the SAD arm of PPT and how do they stratify against adenoma recurrence risk? This is from the sub analysis of the control arm I wrote about earlier:

To summarise the right hand column, folks with no relapse had median HCy around 12.5micromol/l. Those groups with the worst recurrences generally had median HCy over 14micromol/l. Low HCy is "associated", observationally, with lower adenoma relapses.
Of course those who ate the most plants, fibre and general pre-poop had the lowest HCy... Really begs why the trial totally flopped. Something to do with the difference between observational studies and intervention studies I think! There may be no funding for basic science (ask my wife!) but there seems to be plenty for this sort of dogma support BS!
However, within the dogma that HCy is controlled by eating leaves, how bad must it get if you eat almost no plants, with the emphasis on peeled potatoes when you do indulge in vegi-cide, as little fibre as possible and as much animal fat as practical, say >70% of calories?
Well the HCy comes out, when eating to the Optimal Diet, with a median of about 10.7micromol/l. And relatively few plants were murdered (blame Elizabeth) in the process.

No leaves, no fibre, lots of saturated fat. The occasional spud. Please remember that the data from the OD is observational in nature too. You have to guess what the HCy level was pre OD.
Counter intuitive isn't it? But science does that sometimes. Just look at the money spent on supporting the long dead hypothesis that eating leaves is in some way beneficial for humans. There's a powerful need for self justification of the pig's-ear that PPT turned out to be. Depending on your point of view of course!
Peter
"This study failed to show any effect of a low-fat, high-fiber, high-fruit and -vegetable eating pattern on adenoma recurrence even with 8 years of follow-up"
Now, would anyone like to guess how much the PPT cost? I've no idea, but I guess it cost enough that you are not going to accept a result like that after spending all of those tax dollars (thank goodness it wasn't pounds sterling, it's bad enough funding the FSA on this side of the Pond). So there is now an on-going avalanche of subgroup analysis papers flooding out from PPT, many of which are utterly hysterical. Some actually show, by accident, the injury done to many of the people in the intervention arm of the study. From these new papers you would NEVER guess the main trial primary end point was an utter destruction of the hypothesis that flatulence is good for you.
Let's start with that lovely sub group who ate all the dry beans. Upping your beans by 370% appeared to drop your risk for adenoma recurrence to an OR of 0.35 in the intervention arm. This is music to the fibre-ophile ear. The effect is massive. It really should show in observational studies if it is real. It doesn't.
Sub group analysis of the control group, observational only, suggests that contributing to global warming is NOT justified for colorectal cancer prevention. There is no excuse for flatulence. I dunno. Maybe the intervention trial is correct. But don't hold your breath, just your nose.
That last sub group analysis is particularly interesting once you get hold of the full text (many thanks Anna). It only looks at the control arm, so it's observational in nature. They blood sampled folks at the start of the study then counted polyps after four years on whatever shade of the SAD happened to be chosen by people who have had a polyp removed but no diet advice given (ie the lucky ones).
What is topical to me at the moment is that homocysteine (HCy) correlation with polyp recurrence. HCy at enrolment is a very good predictor of polyp recurrence in this group of untutored polyp formers over the next 4 years.
The suggestion, by the authors, is that reducing HCy will reduce adenoma recurrence. Really, someone should test this hypothesis. Maybe a trial increasing fruit, vegetables and fibre should be done to see if these miracle foods will reduce adenoma formation over a four or even eight year period?
Oh, it's been done has it? Ah yes, that's where this data trawl has come from! I remember, yes, that's it, eight years of fruit and fibre does bugger all to prevent polyp recurrence. Silly of me to forget. There was an intervention arm to PPT!
Remember that and re read the very carefully worded abstract... Would you have guessed?
Before I get on to a dietary intervention which might lower HCy (if you care, all this is observational), let's have a look at another subgroup analysis of the PPT data.
I now want to look at what happened in the intervention arm. We can tease out a sub group, the folks who really complied with the intervention, and compare them with the people in the intervention arm who complied poorly.
That's this subgroup analysis.
A total of 210 "super compliers" were noted and compared to controls. I can't really tell if the controls were on the SAD or were those less compliant members of the intervention arm, probably the latter. The odds ratio for adenoma recurrence of supercompliance was 0.65 and statistically significant.
So 210 people out of the 1,905 in the intervention arm benefited. That means that 1,695 people didn't benefit from four years of advice.
But hold on a minute. When the initial study compared intervention with non intervention there was no benefit from intervention compared to the SAD. So if 210 people did better than average, then 1,695 people MUST HAVE DONE WORSE than average. That is, they did worse than those on the SAD. You would have expected a 50:50 split, obviously half should do better and half should do worse than average!
EDIT: Of course if they compared 210 super compliers to the 1,695 injured people they possibly got p < 0.05 because these 1,695 did particularly badly. I wonder if p would have been < 0.05 if they had compared the 210 to the whole SAD group? Probably not would be my guess. Or they would have done it! Me, cynical? Surely not... END EDIT
We can summarise: 1,695 people were injured by being in the intervention arm of the PPT.
I would also point out that "super compliers" are super compliers. These are the sort of people who meticulously take their placebo tablets in clinical trials and consistently do better than those who are lackadaisical with consuming their placebo tablets. Mike Eades has blogged about it nicely here.
So did the 210 super compliers benefit from the diet or were they just the sort of people who comply with authority and automatically do well despite their doctor's best efforts? There was no scope for an adherer effect in the SAD non-intervention arm because they were left alone with no religious group therapy support. Oops, I mean dietary advice...
So was everyone injured in the intervention group but a few had their injury offset by the warm glow of study compliance?
Don't hold your breath waiting to see THAT get discussed in an NIH funded paper. We're looking to justify the cash spent on a superb study which has made no friends among people with control over the purse strings...
Anyhow, back to HCy. We all know from nauseous observational stuff that PPT intervention should have dropped HCy (I've not seen this analysis anywhere yet but I guess it did) but it still didn't drop adenoma recurrence and it probably worsened it in that vast majority of the participants who weren't super compliers.
What sort of HCy levels are present in the SAD arm of PPT and how do they stratify against adenoma recurrence risk? This is from the sub analysis of the control arm I wrote about earlier:

To summarise the right hand column, folks with no relapse had median HCy around 12.5micromol/l. Those groups with the worst recurrences generally had median HCy over 14micromol/l. Low HCy is "associated", observationally, with lower adenoma relapses.
Of course those who ate the most plants, fibre and general pre-poop had the lowest HCy... Really begs why the trial totally flopped. Something to do with the difference between observational studies and intervention studies I think! There may be no funding for basic science (ask my wife!) but there seems to be plenty for this sort of dogma support BS!
However, within the dogma that HCy is controlled by eating leaves, how bad must it get if you eat almost no plants, with the emphasis on peeled potatoes when you do indulge in vegi-cide, as little fibre as possible and as much animal fat as practical, say >70% of calories?
Well the HCy comes out, when eating to the Optimal Diet, with a median of about 10.7micromol/l. And relatively few plants were murdered (blame Elizabeth) in the process.

No leaves, no fibre, lots of saturated fat. The occasional spud. Please remember that the data from the OD is observational in nature too. You have to guess what the HCy level was pre OD.
Counter intuitive isn't it? But science does that sometimes. Just look at the money spent on supporting the long dead hypothesis that eating leaves is in some way beneficial for humans. There's a powerful need for self justification of the pig's-ear that PPT turned out to be. Depending on your point of view of course!
Peter
More fighting talk
Very briefly because there's nothing here we don't know inside out, this is fighting talk. More along the lines of de Lorgeril but in a very public forum.
Does anyone remember how permanent the Berlin Wall seemed for decades and how quickly it fell when it went....
Peter
Does anyone remember how permanent the Berlin Wall seemed for decades and how quickly it fell when it went....
Peter
Sunday, July 11, 2010
Rimonabant and hemopressin
"All right Mr Bender, before we enrol you in this clinical trial I need to be certain that you do not now, or ever have in the past, partake of any recreational drug, specifically those targeting the CB1 receptors in your brain?"
"Well yeah man, like sure, err, I mean, like no, like what was the question?"
"I'll take that as a no"
"Well sure doc, whatever, like, that's cool. You got something new for me to try?"
"We've developed hemopressin, a totally natural appetite modifying peptide. We don't know where it works in the brain, which neuronal circuits it influences or whether it is safe in humans. We think it's a good idea to give it to you at industrial dose rates which will flood your whole body to boost the micromolar concentrations naturally present in your brain. We think it's harmless and won't produce the depression or suicidal ideation which had Rimonabant withdrawn from the market a few years back. There's a big cheque involved."
"Ok doc, whatever. Like, ok, err, how much did you err like err, um, pay me?"
"My secretary will assist you with completing the deposit slip for your bank account. You do have a bank account?"
"Yeah, well, yeah, well, I can't remember really, did you say bank errr account...?"
"That door there, Mr Bender, the one you came in through."
"Ok, yeah, right"
Two weeks later
"Ah, good morning Mr Bender, a lovely day."
"What's the point? I think I'll just kill myself"
"How is you preoccupation with food?"
"What's the point? I think I'll just kill myself."
"Have you noticed any change in post coital satisfaction?"
"What's the point? I think I'll just kill myself."
"How is your satisfaction rating for visual appreciation?"
"What's the point, I think I'll just kill myself."
"Heard any good bands in the last two weeks?"
"What's the point, I think I'll just kill myself."
"Hmmmmmm. But interest in food is really down, yes?"
"What's the point, I think I'll just kill myself. Oh, Doc, before I go do it, those obese babies in nappies, are they on dope to get that obese? And the fat bankers too, they pot heads too? Never mind. What's the point, I think I'll just kill myself."
"Hmmmmm, partial success with that one I think..."
This post might be less perplexing if you've read this press release about this paper. Hemopressin will bomb like Rimonabant. It's based on the "toxic food environment" hypothesis, which says that the only reason I weigh under 65kg is my diet is too non-hedonistic for me to over indulge in. Conversely a toke of dope should make you obese by rendering the contents of the food aisle of a late night garage in to an hedonistic experience....
BTW, being a good member of law abiding citizenry I have no idea who does or doesn't do dope. But obesity is not a feature of any of the people whom I don't know don't do dope. I've heard about the munchies. I guess dope heads must eat less next day or exercise more, the skinny ones that is....
Peter
OK back to the OD and homocysteine as soon as I can get the next post written. Loads of great links have been emailed to me too, if I could just remember where I left them...
"Well yeah man, like sure, err, I mean, like no, like what was the question?"
"I'll take that as a no"
"Well sure doc, whatever, like, that's cool. You got something new for me to try?"
"We've developed hemopressin, a totally natural appetite modifying peptide. We don't know where it works in the brain, which neuronal circuits it influences or whether it is safe in humans. We think it's a good idea to give it to you at industrial dose rates which will flood your whole body to boost the micromolar concentrations naturally present in your brain. We think it's harmless and won't produce the depression or suicidal ideation which had Rimonabant withdrawn from the market a few years back. There's a big cheque involved."
"Ok doc, whatever. Like, ok, err, how much did you err like err, um, pay me?"
"My secretary will assist you with completing the deposit slip for your bank account. You do have a bank account?"
"Yeah, well, yeah, well, I can't remember really, did you say bank errr account...?"
"That door there, Mr Bender, the one you came in through."
"Ok, yeah, right"
Two weeks later
"Ah, good morning Mr Bender, a lovely day."
"What's the point? I think I'll just kill myself"
"How is you preoccupation with food?"
"What's the point? I think I'll just kill myself."
"Have you noticed any change in post coital satisfaction?"
"What's the point? I think I'll just kill myself."
"How is your satisfaction rating for visual appreciation?"
"What's the point, I think I'll just kill myself."
"Heard any good bands in the last two weeks?"
"What's the point, I think I'll just kill myself."
"Hmmmmmm. But interest in food is really down, yes?"
"What's the point, I think I'll just kill myself. Oh, Doc, before I go do it, those obese babies in nappies, are they on dope to get that obese? And the fat bankers too, they pot heads too? Never mind. What's the point, I think I'll just kill myself."
"Hmmmmm, partial success with that one I think..."
This post might be less perplexing if you've read this press release about this paper. Hemopressin will bomb like Rimonabant. It's based on the "toxic food environment" hypothesis, which says that the only reason I weigh under 65kg is my diet is too non-hedonistic for me to over indulge in. Conversely a toke of dope should make you obese by rendering the contents of the food aisle of a late night garage in to an hedonistic experience....
BTW, being a good member of law abiding citizenry I have no idea who does or doesn't do dope. But obesity is not a feature of any of the people whom I don't know don't do dope. I've heard about the munchies. I guess dope heads must eat less next day or exercise more, the skinny ones that is....
Peter
OK back to the OD and homocysteine as soon as I can get the next post written. Loads of great links have been emailed to me too, if I could just remember where I left them...
Sunday, July 04, 2010
Cholesterol hypothesis in 2010 part 2
Brief news update, keep reading the discussion (pdf). Ridker vs de Lorgeril goes on. Jupiter is looking like a gift to truth. Has to happen at some stage! Another great link from O'Primitivo.
EDIT: Here and here are two copies of the original article, just pulled up from comments. One has some problem with a Japanese character set (probably the first).
Peter
BTW comment moderation is in place (sorry Ricardo) on older posts to keep the viagra spam under control. I'll get to genuine comments asap, which is not always that soon at the moment!
EDIT: Here and here are two copies of the original article, just pulled up from comments. One has some problem with a Japanese character set (probably the first).
Peter
BTW comment moderation is in place (sorry Ricardo) on older posts to keep the viagra spam under control. I'll get to genuine comments asap, which is not always that soon at the moment!
Saturday, July 03, 2010
David Blaine and refeeding syndrome
I was looking around pubmed for the effects of fasting on homocysteine concentration. I'd seen a reference which claimed that HCy rose progressively with extended fasting beyond 12h, making a 12h fast the ideal time to measure. For some reason (probably to do with having 10 windows open in two web browsers) I lost it. While pubmeding to relocate it (and failing) I tripped over this rather nice paper on starvation.
It looks very like the second author is the chap in the box and this news report gives an idea of how the first and second authors interacted. It also explains why there were no "pre box" blood values for Blaine!
The paper has a number of gem lines in it. First was that, whatever homocysteine levels do in acute starvation, by 44 days of water fasting they are normal. Normal does not appear to be zero.
Next was the acute hypophosphataemia induced by re feeding. This kicked in by the end of day one. By this time Blaine had been given three cartons of a commercial liquid "food" including a total of just under 40 grams of sucrose. I wouldn't like to mention fructose as a trigger for hypophosphataemia, but it does get an honourable mention here and we've all listened to Lustig discuss fructose the net. Glucose also requires its place in the syndrome too. Dr Powel-Tuck seems to feel that this hypophosphataemia is to do with the energy intake. I suppose this is true if you are stuck in the time warp that energy=glucose, before anyone realised that free fatty acids could be used for energy. As far as I am aware no one has ever precipitated acute hypophosphataemia with an Intralipid infusion, disgusting though the thought of soya bean oil intravenously might be.
You just have to wonder what a supply of calories as fat and protein might have done by perhaps using, gasp, cheese. High in phosphate anyway.... Then add that essential nutrient, sucrose, later. In fact on day five Blaine took himself to the hospital canteen and had fish and chips against doctors orders. Marked fluid retention was the end result but nothing worse.
The fluid retention is very interesting. This is what they noted:
"A raised haematocrit on admission (reflecting haemoconcentration) changed to progressively lower values, despite cautious refeeding, and total avoidance of saline or additional salt during the hospital stay (Table 3). Albumin concentration on admission to hospital was slightly raised and declined gradually over 5 days probably due to plasma expansion. By day 10, 5 days after he left hospital on a free diet, he demonstrated mild pitting oedema."
and here is my favourite
"As refeeding raises plasma insulin, [then] potassium, phosphate and magnesium are driven intracellularly, and sodium extracellularly, expanding circulating volume and causing haemodilution, as indicated in D B by changes in his haematocrit and albumin levels."
It doesn't mention that insulin also causes renal sodium retention, but it does. I rather like this paper, I've not worked through the free full text but I note from the abstract that an insulin infusion with normoglycaemia causes sodium retention even in control rats, never mind spontaneously hypertensive rats. You learn so much from the control groups, even if thay are only rats. And we all thought that folks with hypertension while hyperinsulinaemic on a high carbohydrate diet were born with an in-built frusemide deficiency! Sounds more like Fanta poisoning to me. So, for Blaine, you have to ask whether the immediate return to a "balanced" diet, rich in nourishing sucrose and insulin spiking starch, is what precipitated the fluid retention and whether a more lipid based diet with gradual transition at real food for a few days before diving in to the "sugar as the main course with spuds for dessert" regime might have been less problematic.
I can't imagine anyone testing this idea, so perhaps it's a good job we have phosphate infusions available in UK teaching hospitals to pull post-starvation patients back from the brink of iatrogenic sugar poisoning during refeeding!
Peter
It looks very like the second author is the chap in the box and this news report gives an idea of how the first and second authors interacted. It also explains why there were no "pre box" blood values for Blaine!
The paper has a number of gem lines in it. First was that, whatever homocysteine levels do in acute starvation, by 44 days of water fasting they are normal. Normal does not appear to be zero.
Next was the acute hypophosphataemia induced by re feeding. This kicked in by the end of day one. By this time Blaine had been given three cartons of a commercial liquid "food" including a total of just under 40 grams of sucrose. I wouldn't like to mention fructose as a trigger for hypophosphataemia, but it does get an honourable mention here and we've all listened to Lustig discuss fructose the net. Glucose also requires its place in the syndrome too. Dr Powel-Tuck seems to feel that this hypophosphataemia is to do with the energy intake. I suppose this is true if you are stuck in the time warp that energy=glucose, before anyone realised that free fatty acids could be used for energy. As far as I am aware no one has ever precipitated acute hypophosphataemia with an Intralipid infusion, disgusting though the thought of soya bean oil intravenously might be.
You just have to wonder what a supply of calories as fat and protein might have done by perhaps using, gasp, cheese. High in phosphate anyway.... Then add that essential nutrient, sucrose, later. In fact on day five Blaine took himself to the hospital canteen and had fish and chips against doctors orders. Marked fluid retention was the end result but nothing worse.
The fluid retention is very interesting. This is what they noted:
"A raised haematocrit on admission (reflecting haemoconcentration) changed to progressively lower values, despite cautious refeeding, and total avoidance of saline or additional salt during the hospital stay (Table 3). Albumin concentration on admission to hospital was slightly raised and declined gradually over 5 days probably due to plasma expansion. By day 10, 5 days after he left hospital on a free diet, he demonstrated mild pitting oedema."
and here is my favourite
"As refeeding raises plasma insulin, [then] potassium, phosphate and magnesium are driven intracellularly, and sodium extracellularly, expanding circulating volume and causing haemodilution, as indicated in D B by changes in his haematocrit and albumin levels."
It doesn't mention that insulin also causes renal sodium retention, but it does. I rather like this paper, I've not worked through the free full text but I note from the abstract that an insulin infusion with normoglycaemia causes sodium retention even in control rats, never mind spontaneously hypertensive rats. You learn so much from the control groups, even if thay are only rats. And we all thought that folks with hypertension while hyperinsulinaemic on a high carbohydrate diet were born with an in-built frusemide deficiency! Sounds more like Fanta poisoning to me. So, for Blaine, you have to ask whether the immediate return to a "balanced" diet, rich in nourishing sucrose and insulin spiking starch, is what precipitated the fluid retention and whether a more lipid based diet with gradual transition at real food for a few days before diving in to the "sugar as the main course with spuds for dessert" regime might have been less problematic.
I can't imagine anyone testing this idea, so perhaps it's a good job we have phosphate infusions available in UK teaching hospitals to pull post-starvation patients back from the brink of iatrogenic sugar poisoning during refeeding!
Peter
Folate and homocysteine
Anna (many thanks) supplied me with a copy of the paper with a title which suggested that homocysteine was a stimulator of sulphate incorporation in to cartilage comparable to ILGF-1 (somatomedin/sulphation factor in those days). They were looking at concentrations of homocysteine comparable to those seen in children with the genetic lack of the enzyme cystathione synthetase, around 10mg/dl. That's VERY high, about 10 times the level in the blood of a normal person.
At this concentration there are undoubtedly marked skeletal deformities in children with homocysteinuria, presumably due to elevated homocysteine mimicing ILGF1 in their growing bones. As normal children appear to develop GAG containing arteriosclerosis at an early age then the ability of markedly elevated homocysteine to produces excessive GAG incorporation in to cartilage might very well be expected to increase the incorporation of GAG in to arterial plaque from physiological amounts to pathological amounts. That doesn't seem controversial to me.
The question is then whether homocysteine, at the concentrations seen in the plasma of the genetically more normal general population, drives arteriosclerosis. And, much more interesting, whether lowering homocysteine prevents cardiovascular disease in this population.
The vast majority of the work on homocysteine was done by Kilmer McCully and is covered by his book The Homocysteine Revolution. I think it's reasonable to summarise his approach to diet and cardiovascular health as to "eat Food". Food, with a capital F, denotes sources of B6, B12 and folate, three of the main factors for the metabolism of homocysteine. Needless to say there is a marked de-emphasis of sucrose and white flour consumption. But there is also a de-emephasis on white fat. Obviously there is not a whole lot of B6, B12 or folate in lard or beef dripping. When you take this line of thought to its logical conclusion you end up with a diet containing lots of fruit, vegetables, fresh whole grains and good quality lean meat. It's not exactly the Optimal Diet, in fact the term that comes to mind is "Ornish" plus meat. Would this work? Who knows? As far as I am aware Ornish has never tested his diet ideas. But the concept of whole food, freshly prepared, makes a lot of sense provided you don't mind the auto immune diseases from gluten and have not already broken you liver with decades of the SAD to allow blood glucose of spike to 200mg/dl after a bowl of whole meal pasta.
For folks too lazy to cook or juice some leaves, how about a B12/folate/B6 pill? Well the Norwegians have checked it and the initial problem is that it doesn't work for CVD, though it drops homocysteine nicely. The paper is from 2009. You can read the full text if you would like the to know their proposed mechanisms for the failure of vitamin induced reductions of homocysteine to prevent CVD events, but here's the executive summary:
"All the above mechanisms partly explain why clinical trials on Hcy lowering, such as the VISP, NORVIT, or HOPE-2 and possibly others to be conducted, failed to report any improvement in CVD risk and clinical outcome. Maybe more efficient ways to target Hcy metabolism, other than vitamin supplementation, should be examined. Focusing on increasing Hcy renal clearance or reducing asymmetric dimethylarginine levels would be potentially useful. In addition, other strategies able to increase intracellular 5-MTHF should be developed, to achieve the maximum regulation of intracellular Hcy metabolism."
The problems with vitamin treatment for elevated homocysteine in CVD have continued to in 2010. There has also been a follow on report on cancer and all cause mortality, a 2010 paper from the Norway studies, which does not make the lowering of homocysteine by using B vitamins look too helpful.
The increase in all cause mortality is a very interesting finding. You could argue that, in people without genetic homocysteinuria, that there might be some benefit from homocysteine at around 13micromol/l which is lost by lowering it to 9micromol/l, assuming you are eating a diet which has already established your heart disease. Or, equally, you could argue that taking vitamin B12 and folate, in the form of a vitamin supplement, is bad for you. Perhaps 0.8mg of synthetic drug grade folic acid is not quite the same as eating a few leaves. Quite a lot of leaves in fact.
The later idea is interesting as the WHEL intervention study, with it's significantly increased vegetable (and so probably folate) intake, didn't actually kill anyone beyond the death rate in the control group. Mind you, it didn't help anyone either but then eating leaves for health is a bit of a weird idea to me.....
So there's the choice. Is B12/folate supplementation bad for you? Or is homocystene helpful in small amounts and damaging in large amounts? A bit like oxygen.
Peter
EDIT there has been a similar report from Oxford with similar trends but with no statistical significance. The Oxford study used 0.4mg folic acid and the placebo group will have been eating UK breakfast cereals which are folate supplemented. Norway does not supplement it's food supply with folate so compared 0.8mg folate with zero supplementary folate level for the placebo group. There looks to be a dose response rate to folic acid toxiciy, if that's what the driver is for the problems. Heartwire produced a nice summary slide.

I realise that none of the changes are statistically significant but every parameter was worse in the folic acid/B12 group. The Norway study is in a better position to find significant effects and p does drop below 0.05. The Norway study is also much less likely to be drug money influenced. Tends to be convincing.
At this concentration there are undoubtedly marked skeletal deformities in children with homocysteinuria, presumably due to elevated homocysteine mimicing ILGF1 in their growing bones. As normal children appear to develop GAG containing arteriosclerosis at an early age then the ability of markedly elevated homocysteine to produces excessive GAG incorporation in to cartilage might very well be expected to increase the incorporation of GAG in to arterial plaque from physiological amounts to pathological amounts. That doesn't seem controversial to me.
The question is then whether homocysteine, at the concentrations seen in the plasma of the genetically more normal general population, drives arteriosclerosis. And, much more interesting, whether lowering homocysteine prevents cardiovascular disease in this population.
The vast majority of the work on homocysteine was done by Kilmer McCully and is covered by his book The Homocysteine Revolution. I think it's reasonable to summarise his approach to diet and cardiovascular health as to "eat Food". Food, with a capital F, denotes sources of B6, B12 and folate, three of the main factors for the metabolism of homocysteine. Needless to say there is a marked de-emphasis of sucrose and white flour consumption. But there is also a de-emephasis on white fat. Obviously there is not a whole lot of B6, B12 or folate in lard or beef dripping. When you take this line of thought to its logical conclusion you end up with a diet containing lots of fruit, vegetables, fresh whole grains and good quality lean meat. It's not exactly the Optimal Diet, in fact the term that comes to mind is "Ornish" plus meat. Would this work? Who knows? As far as I am aware Ornish has never tested his diet ideas. But the concept of whole food, freshly prepared, makes a lot of sense provided you don't mind the auto immune diseases from gluten and have not already broken you liver with decades of the SAD to allow blood glucose of spike to 200mg/dl after a bowl of whole meal pasta.
For folks too lazy to cook or juice some leaves, how about a B12/folate/B6 pill? Well the Norwegians have checked it and the initial problem is that it doesn't work for CVD, though it drops homocysteine nicely. The paper is from 2009. You can read the full text if you would like the to know their proposed mechanisms for the failure of vitamin induced reductions of homocysteine to prevent CVD events, but here's the executive summary:
"All the above mechanisms partly explain why clinical trials on Hcy lowering, such as the VISP, NORVIT, or HOPE-2 and possibly others to be conducted, failed to report any improvement in CVD risk and clinical outcome. Maybe more efficient ways to target Hcy metabolism, other than vitamin supplementation, should be examined. Focusing on increasing Hcy renal clearance or reducing asymmetric dimethylarginine levels would be potentially useful. In addition, other strategies able to increase intracellular 5-MTHF should be developed, to achieve the maximum regulation of intracellular Hcy metabolism."
The problems with vitamin treatment for elevated homocysteine in CVD have continued to in 2010. There has also been a follow on report on cancer and all cause mortality, a 2010 paper from the Norway studies, which does not make the lowering of homocysteine by using B vitamins look too helpful.
The increase in all cause mortality is a very interesting finding. You could argue that, in people without genetic homocysteinuria, that there might be some benefit from homocysteine at around 13micromol/l which is lost by lowering it to 9micromol/l, assuming you are eating a diet which has already established your heart disease. Or, equally, you could argue that taking vitamin B12 and folate, in the form of a vitamin supplement, is bad for you. Perhaps 0.8mg of synthetic drug grade folic acid is not quite the same as eating a few leaves. Quite a lot of leaves in fact.
The later idea is interesting as the WHEL intervention study, with it's significantly increased vegetable (and so probably folate) intake, didn't actually kill anyone beyond the death rate in the control group. Mind you, it didn't help anyone either but then eating leaves for health is a bit of a weird idea to me.....
So there's the choice. Is B12/folate supplementation bad for you? Or is homocystene helpful in small amounts and damaging in large amounts? A bit like oxygen.
Peter
EDIT there has been a similar report from Oxford with similar trends but with no statistical significance. The Oxford study used 0.4mg folic acid and the placebo group will have been eating UK breakfast cereals which are folate supplemented. Norway does not supplement it's food supply with folate so compared 0.8mg folate with zero supplementary folate level for the placebo group. There looks to be a dose response rate to folic acid toxiciy, if that's what the driver is for the problems. Heartwire produced a nice summary slide.

I realise that none of the changes are statistically significant but every parameter was worse in the folic acid/B12 group. The Norway study is in a better position to find significant effects and p does drop below 0.05. The Norway study is also much less likely to be drug money influenced. Tends to be convincing.
Subscribe to:
Posts (Atom)
