Back to the paper provided by met4health, briefly mentioned previously:
Electron Transport Chain-dependent and -independent Mechanisms of Mitochondrial H2O2 Emission during Long-chain Fatty Acid OxidationIt's a very interesting paper. It brings to light some of the problems of using isolated mitochondrial preparations. The basic summary is that if you compare the oxidation of palmitoylcarnitine to either pyruvate/malate or glutamate/malate there is significant ROS generation with palmitate, even at low delta psi, compared to the primarily NADH generating substrates (which produce zero ROS at 180mV delta psi in these preparations).
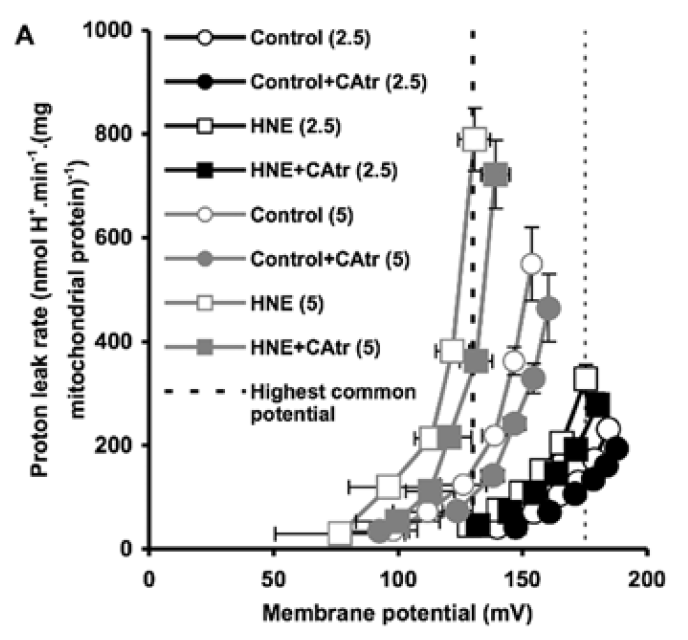
Figure 3 is perhaps the most interesting. For section C they blocked the function of ATP synthase with oligomycin to raise delta psi and then titrated delta psi downwards by uncoupling with FCCP to give either a low or high delta psi. Then they fed the preparation with either palmitoylcarnitine (plus extra carnitine) or glutamate/malate.
The right hand bar graph is derived from the left hand curve and shows that delta psi has some influence but, under both delta psi conditions, there are many more ROS produced under palmitate oxidation than G/M. Delta psi clearly has an effect but substrate also has a marked influence.
That's the convincing part of the paper. There is no insight as to mechanism but my biases assume it will be F:N ratio related. I might have left it there but I can't.
The rest of the results are hugely influenced by this statement from the methods:
"Unless otherwise stated, determinations were made in the presence of oligomycin (3µg/ml) to inhibit ATP synthesis, a condition used in previous studies of the mechanisms of ROS formation in isolated mitochondria and permeabilized muscle fibers (e.g. see Refs. 4, 13, 34, and 38)."
Translation: We blocked ATP synthase because everyone does it.
So pretty well all of the results appear to have been produced during a complete blockade of ATP synthase.
These preparations are always looking at ROS generation when the only dissipation route for membrane potential is some form of uncoupling (UCPs, NNT, various proton assisted co-transporters)
There is no other way to allow O2 consumption under oligomycin. See
last post on Brand's review.
This begs the question of how is it possible to feed a mitochondrial preparation supra maximal amounts of NADH substrates (pyruvate/malate) consuming relatively large amounts of oxygen in the absence of any way of dissipating the proton gradient across the inner mitochondrial membrane without ATP synthase being active?
The answer is that there must be some sort of uncoupling going on. The delta psi of 180mV is completely normal but this does not mean that there is no proton leak through the inner mitochondrial membrane. It merely means that the leak (defined by an O2 consumption of 21nmol O/min/mg of mitochondrial protein) is sufficient to avoid raising delta psi to massive levels in the face of a supra maximal supply of pyruvate/malate. The proton leak is also kept low enough not to drop delta psi. This smacks of regulation.
By comparison palmitoylcarnitine at 18micromol/l has less oxygen consumption and supports a lower delta psi, in the region of 145mV.
This is clear in section A of Figure 3.
Both of these values for O2 consumption under oligomycin *have* to be facilitated via uncoupling.
What is also clear is that, with oxygen consumption lower under palmitoylcarnitine, there is less uncoupling than for P/M.
Has anyone noticed that in Figure 3 there is a flick between 18µM palmitoylcarnitine and 18µM palmitoylcarnitine plus 2mM carnitine between various graphs?
The authors of the paper considered that, with palmitoylcarnitine, the low delta psi combined with low O2 consumption might be due to an inhibitory effect of fatty acid oxidation intermediates on either FAO itself or on ETC function.
Adding 2mM carnitine appears to remove such intermediates. I've not been in to the chemistry but it looks like the carnitine exports them from the mitochondria. There's something about this in the supplementary data. I'm just accepting it happens for today. About which I'm a little cautious.
Adding the extra carnitine makes the oxidation of palmitoylcarnitine look just like supra maximal P/M or G/M.
Here's the oxygen consumption bar chart from supplementary data Figure 3. We're looking at the left hand pair. White bar is palmitoylcarnitine 18µM consuming (as before) 10nmol O/min/mg. Black bar is after the extra carnitine was added. Oxygen consumption is around 25nmol O/min/mg (and delta psi did the same) and is now comparable across the metabolic substrates, black bars:
This increase in O2 consumption means that, under oligomycin, that uncoupling has markedly increased and is directly equivalent to supra maximal NADH sources.
To me this implies that normal fatty acid oxidation (ie without extra carnitine) is a self limiting process.
If the paper is correct (don't forget they are working with oligomycin blocked preparations) a high delta psi is not a feature of palmitoylcarnitine oxidation. The oxidation of palmitoylcarnitine suports ROS generation irrespective of delta psi. To make me really happy it would be nice to generate ROS with different fatty acids and look at the effect of the F:N ratio on ROS generation.
There are certain implications to these thoughts. First is that physiology is very keen to keep delta psi in the region of 180mV or lower and applies some degree of uncoupling to achieve this. This appears to be independent of fatty acid induced uncoupling.
This is possibly very important. Any supra maximal supply of substrate in a non-phosphorylating mitochondrial prep (state 4oligomycin) has to have a method to stabilise delta psi at around 180mV. How? Another post there.
Second is that FAO intermediates down regulate the ETC performance directly, limiting delta psi to 145mV from palmitoylcarnitine 18µM unless those FAO intermediates are removed.
Finally, FAO appears to generate ROS moderately independently of delta psi. Certainly palmitoylcarnitine does.
Now for a long-time-ago throwback:
Permeablised muscle fibres behave pretty much like mitochondrial preparations.
"Despite an increase in whole-body fat oxidation, we observed an overall reduction in both coupled state 3 respiration and maximally uncoupled [here using FCCP] respiration in permeabilized skeletal muscle fibers..."
The RCR using an uncoupler and oligomycin, ie state 3FCCP / state 4oligomycin, fell markedly with fasting, hence the term "skeletal muscle mitochondrial dysfunction" in the title.
But 60 hours of fasting cannot possibly destroy your mitochondria. People can pushbike hundreds of kilometres over 5 days without eating anything at all. Their mitochondria work.
I would suspect that this is a fully physiological control system designed to cope, at the mitochondrial level of fatty acid oxidation, with a potentially limitless supply of energy from the fatty acids released from adipocytes under low insulin/insulin signalling conditions.
In this study using permeablised muscle fibres you could probably have reversed the effect completely by treating with 2mM carnitine.
But why would you want to? Apart from gaining insight as to what is normal physiology of course.
Summary: fatty acid oxidation is a self regulating system at the level of beta oxidation rather than at the level of the Krebs Cycle. I suspect that the FAO intermediates will act directly on the electron transport chain.
If this is correct it will provide insight in to other elevated FAO conditions, ie obesity with insulin resistance, where fatty acids should modify (appropriately) ETC function to avoid energetic overload.
I've had suspicions that this has to be the case for a long time. Finally I'm getting to see a little progress.
Peter