This is another non-referenced, thinking out loud post which is the precursor to more normal technical posts. Here we go.
Too few ROS.
This post is about how generating too few ROS in mitochondria generates excess ROS in those said mitochondria and what physiology does about this.
Here's the stripped down doodle of the electron transport chain I'm going to use

I've left out complex II, cytochrome C etc to keep it very simple. It's not complete, it's a minimal mental "model". You have been warned.
Next is the normal electron flow from NADH and FADH2 to oxygen:

Electrons passing through complexes I, III and IV pump protons out of the mitochondria to produce an electrical and pH difference between inside and out, the mitochondrial membrane potential, delta psi

and of course delta psi is used to generate ATP by ATP synthase, much as a rotating water mill uses hydrostatic pressure to generate usable energy.
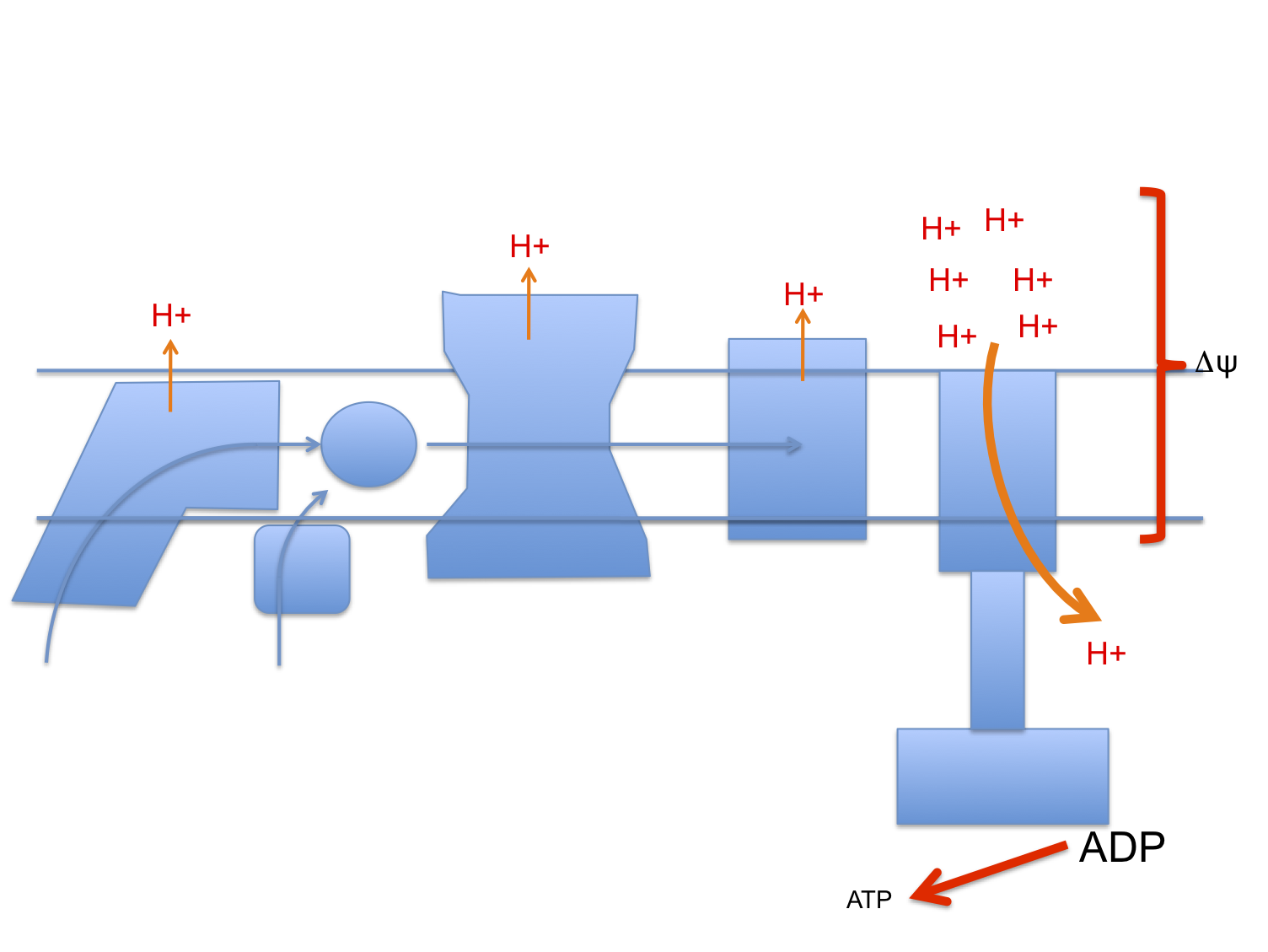
One crucial necessity to allow ATP synthase to function is a supply of ADP. If all of a cell's supply of phosphorylated adenine is in the form of ATP there is minimal ADP as substrate for ATP-synthase to act on, so delta psi will become larger as pumped protons accumulate on the outside of the mitochondrial membrane.

If ATP predominates the cell is replete and the correct response is to limit further caloric ingress. As ETFdh tries to transfer electrons on to the CoQ couple and subsequently to complexes III and IV it becomes progressively harder to pump protons against a rising delta psi. At some point it becomes easier to divert electrons from ETFdh through complex I as reverse electron transport (RET) which generates a very specific, localised ROS signal which is designed to limit insulin signalling and be quenched using superoxide dismutase without doing damage. Saturated fats produce this signal very well because their lack of double bonds maximises the input of FADH2 to facilitate RET:

In this mental model palmitate limits caloric ingress, stops excessive proton pumping and maintains delta psi within physiological limits.
Next we have the situation under linoleic acid oxidation. Here there is a reduced input of FADH2 from ETFdh so it is more difficult to generate the RET needed to limit caloric ingress when ATP is replete and ATP-synthase is no longer consuming delta psi. Protons continue to be pumped and delta psi rises to supra-physiological levels

As delta psi rises the ability to generate RET through complex I also rises until eventually even the relatively low FADH2 input from linoleate can produce RET. However this high delta psi will also allow the generation of ROS at multiple sites in the ETC in addition to that at complex I. Complex IV seems to be a minimal site for this but complex III will produce ROS from sites facing both the mitochondrial matrix and the cytoplasm while complex I appears to only generate on the matrix side, probably from multiple sites under very high delta psi. ETDdh (and mtG-3-Pdh and complex II) can also generate ROS under high delta psi conditions:

This is both good and bad.
Good because it finally hits the signalling pathways needed to limit caloric ingress. Bad because it hits lots of other components of the cell structure in addition.

Summary: consuming linoleic acid will cause your mitochondria to explode.
Except that's preposterous, they don't.
The simplest protective measure is the diversion of calories within the cell in to storage. Those calories are only present in the cell secondary to excess insulin signalling. Because diversion to storage is a classical function of insulin signalling, this will drive obesity whilst also providing some protection from pathological ROS generation.
Additional protection comes from uncoupling.
The core mechanism for the generation of excessive ROS under unmitigated LA oxidation is high delta psi.
The core mechanism for the generation of excessive ROS under unmitigated LA oxidation is high delta psi.
Uncoupling lowers delta psi. Doing this is all that is necessary to protect against pathological ROS generation.
But wait.
If linoleic acid allows excess caloric ingress due to a deficit in physiological ROS generation, surely non-specific suppression of ROS generation should increase caloric ingress, increase delta psi to overcome the degree of uncoupling present and re establish pathological ROS generation? Alternatively might uncoupling go on to allow even more excess calorie storage?
Neither happens.
I'll run through some of the papers I've been looking at over the last few months and post about them next.
Peter

25 comments:
This post reminded me of an excellent presentation given by Mike Eades in 2018. 'A New Hypothesis of Obesity' The technical part starts at 22mins in.
https://www.youtube.com/watch?v=pIRurLnQ8oo
Yes, that post was based on Peter's work, Eades mentions that.
"Except that's preposterous, they don't."
They don't. They implode. Destroying the cristae does a fine job of breaking the ETC and preventing that explosion...
"A novel observation in this study was abnormal condensed mitochondria, but only in the PD group (Fig. 5B)."
"What's Worse—Carbs or Seed Oils? Understanding a High-PUFA Diet."
http://yelling-stop.blogspot.com/2018/06/whats-worsecarbs-or-seed-oils.html
Tucker, yes, the PUFA/streptozotocin study, a good one. Complete caloric overload in the near absence of insulin. If caloric exposure is massive then pathological ROS will be unstoppable, especially if acute in onset.
I have another interesting (to me) aside in another study where they injected oligomycin repeatedly in to one knee of a group of rats. I can't imagine a better way of generating ROS than totally disabling ATP synthase. I'm guessing the peri-articular cells didn't die acutely as they switched to glycolysis but the inflammatory reaction was impressive and makes me think very strongly about ROS from high delta psi. As in CVD is an "inflammatory" disease/process which we don't treat with corticosteroids... More ways than oligomycin to disable the ETC.
Peter
Quite OT, so ignore if you like: have you run across anything in the literature explaining what's going on metabolically, when diabetics get that putrid-honey/fruity-corpses smell? I'm keen to know what's happening chemically, but can't find any explanation-- the internet just spits out the same answer over and over again: it happens with DKA. Well, yes, but *what*, precisely, is happening? Do we actually know what chemical processes are involved with that, or is it something science hasn't investigated? Inquiring minds want to know... I've seen (or rather smelled) it happen over and over in the apparent absence of high glucose, so I'm wondering if understanding the process that causes it might give more insight into what's going on in that circumstance.
Any insights?
That's the smell of ketones. They warn if you go on a keto diet the same thing could happen (never happened to me). This article might be helpful: https://www.ruled.me/what-is-acetoacetate/
"Once acetoacetate [a ketone body] is created, it will have one of three fate: (1) It’ll be sent off to other cells and used as fuel, (2) it’ll be converted into a more energy efficient ketone body called BHB and used as energy throughout the body, or (3) it will spontaneously react to create acetone (which is excreted — typically through the breath — and is not used for energy)."
Hi JustPeachy,
No, I've not considered this. As a pointer I would suggest that neither ordinary DMT2 or DKA as primarily about glucose. They are about lipids. We've mentioned spoilage smells in junk food in the past and Tucker pointed out that these tend to be omega 3 based rather than omega 6 based. Might be somewhere to go looking.
Peter
@cavenewt: I've pondered this, and what you're talking about is not the same smell. I have an alarmingly keen nose, and my husband (who has a metabolism like a blast furnace) often gets the acetone smell when fasting. It is a distinctly astringent chemical kind of smell.
The fruity smell is different (and differently revolting!). From a purely non-scientific, mostly-aesthetic perspective, it smells like the body's trying to sweat out excess sugar, through rotting skin. Just horrifying.
@Peter: thanks for the pointer. Chemistry's not my strong suit, but I'll poke around and see if I can find anything. I'm anxious to understand it, because I'm a T2D (heavily familial, alas) controlling with diet and exercise. I got a little loosey-goosey with diet and tracking over the holidays, and was alerted to the problem by waking up with "the diabetes smell" (yuck!) and a fasting glucose of 120 (US units). I'm back on track now, not registering *any* high numbers, though fasting AM still in the 90s (I'd like them to be in the 80s)... but I'm still getting the DKA smell, off and on. It is driving me nuts, because to me it's an indicator that *something's wrong*, but I can't interest a doctor because the numbers are fine. Sigh. If I could just understand the why/how of it...
People look at glucose because its elevation the canonical definition of diabetes. But diabetics also have elevated FFAs. If those FFAs have plenty of double bonds who knows what derivatives they may form....
Peter
@Peter: Clearly, I have some homework to do.
JustPeachy re: " it smells like the body's trying to sweat out excess sugar". I am T2D also with a multi generational familial history of it. I have often wondered though if family food culture also has something to answer for. Anyway, mine came to a head about 18 years ago and at the time I discovered just how high my bg had become (17mmol/l, a lot!) I had also been experiencing a lot of skin rashes, oiliness, odours and so forth and I realised this probably was sugar working it's way out and feeding up various populations of skin bacteria. It wasn't nice. I've kept my bg near normal levels since then and my skin is as clear as it was as a child.
Peter, very interesting about FFA which would explain the oiliness. Perhaps it's also the case that the sense of taste and smell is affected by the internal chemical imbalances.
@Passthecream: Honestly, I'm baffled. I mean, I woke up with the DKA smell this morning. Checked my blood sugar: 84 (4.7mmol/l)-- rock solid totally normal fasting number. It was 92 (~5.0mmol/l) before I went to bed. I probably ate fewer than 20g of carbs all day yesterday. Everything looks normal and fine (and I worked hard to get there!) so WHY do I wake up smelling like a metabolic emergency?? I would love to write that off and say "nothing to see here" but A) It's intermittent, B) sometimes it really is a warning sign that my numbers are creeping up and I need to get myself back on the low-carb/obsessive-monitoring wagon, and C) a 24-72 hour fast *always* makes it go away. Oh, and D) it almost always happens overnight.
If it's sugars working their way out through my sweat (and it could be! I think.), my best guess is that my system has an extraordinarily low threshold for dumping sugar into my sweat glands, and/or my liver and pancreas are playing some wacky game while I sleep, releasing boatloads of sugar into my bloodstream and then packing it away again before morning. I have never, to my knowledge, been anywhere near actual DKA territory.
I've no doubt that genes, prenatal environment, *and* poor childhood diet have a lot to do with the T2D: My mom, my maternal grandmother, and my maternal great-grandmother were *all* T2Ds (and none were ever obese!)(I am relieved that I have only sons, and that mitochondrial line ends with them). Then, to top it off, my parents were junk-food vegetarians, so you know: a childhood of margarine, cheerios, instant oatmeal, and stir-fried tofu, followed by the inevitable PCOS and T2D. Low-carb has worked really well to keep things under control (some lapses, alas). I've never been more than 30 pounds overweight... but the first time I noticed that horrible smell, I was only 20 years old, and within the healthy weight range. That was before cheap glucometers so I've no idea what my sugars were doing, but the first obvious signs of PCOS showed up at about the same time so... it's related.
The main question for me is... I know there's some relation between the smell and my metabolic state, and that means *it's information*. It's telling me something. I just want to know *what it's telling me*.
Do you know this interesting study?
https://www.sciencedirect.com/science/article/pii/S0021925819375520
It looks like superoxide production from fatty acid oxidation is not much influenced by membrane potential. It is produced even when potential is low. I think this can differentiate between fat and glucose as signal. I wonder if DECR enzyme and NADP+ signalling could better explain the main link between PUFA and chronic diseases. Eg when DECR is knocked down in mice it disables gluconeogenesys, why? And the similarity with polyol pathway effect on NADPH/NADP+? I'm only layman but isn't it interesting?
Hi met4health,
Thank you. That's a keeper. I've been looking for something along those lines for many years. It's quite clear that under extended fasting with FFAs over 2000microM that there is s control system in place to limit unrestrained ATP generation. The paper has lots of detail and leads.
Many thanks
Peter
Peter, this is lovely stuff and nice to see those shapely doodles again. However I am still struggling to understand how Metformin might fit into these squiggles. As an mth3pdh inhibitor you'd expect a partial inhibition of etc input at ETFdh, no? Why doesn't it lead to greatly increased ros leakage leading to damage in some contexts eg a business as usual SAD??? Or does it? I can see how it would lower insulin resistance in the same way that pufa do --- innapropriately. Is it a ' bad thing really?
Here's an interesting case study of a (genetic, homozygous autosomal recessive snp) Lipid storage myopathy caused by dysfunctional EFTdh:
"Clinical characteristics and gene mutation analysis of an adult patient with ETFDH-related multiple acyl-CoA dehydrogenase deficiency"
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7533516/
"Muscle biopsy specimens exhibited a significant increase of the lipid droplets within the muscle fibers, which was consistent with the pathological characteristics of lipid storage myopathy. In addition, a few scattered atrophic myofibers were observed, 12–30 µm in diameter. Small vacuoles in uniform size were observed in several muscle fibers, while some vacuolar muscle fibers were slightly basophilic. Furthermore, ORO staining revealed a massive accumulation of lipid droplets within the vacuolar muscle fibers, with some of them fused into sheets."
That leads me to a paradoxical position. 1) don't take Metformin while following a regular high carb diet. It will lead to inappropriate insulin sensitivity. (Standard conditions apply.) 2) Don't take Metformin on a high fat diet - excess lipid will accumulate .
Pass, I'm not sure how metformin inhibiting mtG3Pdh would impact on ETFdh... Also the lipid in the vacuoles does not have to be triglyceride (ie esterified by insulin). It's possible the lipids are simply acyl-CoA derivatives from beta oxidation in free form. The fact that they stain with ORO just means they are fatty/oily, not that they are normal triglycerides...
I think metformin works very much like uncoupling agents. Its direct effect is as an inhibitor on insulin signalling (the poor girl in Poland tinyurl.com/bdet833j). It's the slim phenotype produced by resisting insulin which is insulin sensitive, once the metformin has worn off.
Metformin has the added complication of resisting the effect of exposure to high insulin which should produce the normal insulin-induced insulin resistance in the acute phase. As in tinyurl.com/4zsvznpy
So. metformin inhibits normal lipid storage by "normal" levels of insulin. "High" insulin levels cause insulin resistance. Metformin lowers insulin signalling enough to block this.
Both effects are by blocking insulin induced ROS at mtG3Pdh. Or so I think...
Peter
Peter, ta.
I think I'm getting a complex complex.
It matters a lot if you mean the Redox status of beta cells versus the Redox status of the destination cells, the insulin consumers. Me, thinking something like the latter, Metformin lowers mt Redox status via fad bound mth3pdh ( at sdh location??) ---> less insulin resistance under high delta psi conditions similar to the effect of pufa. Seemingly however, ETFdh has some extra leverage over this process???
That's spellcheck putting an h instead of a g. But I've been known to be wrong many many many times before :/
Hi Pass, excellent reasoning. This has, in the past, driven me slightly crazy. Or maybe I should say crazier.
Just ignore the insulin-induced insulin resistance aspects to keep it simple for now (ask if you want to go there).
Metformin should produce a reduced insulin/glucose response similar to the partial insulin gene knockout mice in Jim Johnson’s lab. In early life those mice have some degree of glucose intolerance but by adulthood they are slim, insulin sensitive and normoglycaemic secondary to longterm reduced insulin exposure. They should be diabetic, but their maintained insulin sensitivity from low insulin exposure stops this.
Metformin imitates this but is generally given to adult obese humans, so it should produce a functional shift towards a diabetic phenotype.
However metformin, in addition to limiting insulin’s secretion and action, also suppresses hepatic glucose output. This appears to be independent of insulin signalling. It can be claimed to be an effect via mtG3Pdh:
Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4074244/
but this is arguable!
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4154964/
However, whatever the mechanism, metformin *does* suppresses hepatic glucose output and the reduced glucose output *will* limit the hyperglycaemia which *should* occur acutely under low insulin secretion/signalling conditions in an adult obese human.
PUFA also limit insulin secretion (as they do in isolated perfused pancreas preps) but do nothing to suppress hepatic glucose output, so plasma glucose should rise and, in an intact organism, this hyperglycaemia will restore glucose stimulated insulin secretion leading to restored insulin levels combined with insulin sensitised hepatocytes (and adipocytes) with subsequent fatty liver/weight gain.
It took me an embarrassingly long time to realise the importance of decreased hepatic glucose output under metformin.
Peter
Thanks Peter. Good clarification. Also reassuring to think I'm in good company!
Re-reading that paper where they elucidated the action of Metformin to inhibit mtg3pdh, they mention varying results amongst the previous research depending on dosage plus starting point of the subjects. I was surprised at their definition of a therapeutic dose - 50 to 200mg/Kg. That minimum is approx four times any dose I ever took. Horrible bitter stuff it was too.
Hi Peter,
I return to may be supplemental or alternative theory using DECR enzyme as tool, because it looks like nobody is interested, I coudn't find any mention on your or similar blogs. But it's enzyme used only for PUFA beta oxidation, not for SFA nor MUFA. And it generates NADP+.
https://en.wikipedia.org/wiki/2,4_Dienoyl-CoA_reductase
I wonder if NADP+ production is'nt proxy for rate of all fats burning - signal for elevating gluconeogenesys through Ca+ signal. NADP+ is immediately converted to NADPH but by different pathways. It reflects rate of GSSG reduction to GSH, so rate of neutralizing H2O2 from mitochondria and peroxizomes. Burning PUFA makes this signal excessive and false, I guess that could be 10x more intensive signal from PUFA than from other fats. Only about 1% of O2 is converted to O2- and to H2O2 from SFA or MUFA, but each molecule of LA makes NADP+. So 1% of PUFA, no problem, but 10% has to have consequences, don't you think?
Just only an idea, because lack of FADH2 looks fine, but I see it as too small signal to cause such big metabolic effects. Thank you for your work.
Jaromir
Hi Jaromir,
Have you looked at Brad Marshall's ideas at https://fireinabottle.net/?
I would also make plain that the F:N ratio from isolated fatty acids is also a "mental model". A starting point for thinking. Once you get on to mixed lipids all sorts of things become apparent. Also that concept that ROS can be generated without high delta psi goes back to FADdh generating ROS in proportion to the FADH2 being presented (if I read correctly, not had much time for the detail yet).
Pass, they’re rats and mice, to achieve a given plasma level you need much higher oral doses than for larger body surface area animals, you don’t work by weight. I tend to believe rodent studies using oral doses which don’t kill the model animal. That’s in stark contrast with cell culture using 1000microM when plasma conc of 200microM puts a human in the ICU with no guarantee of coming out alive.
Peter
Yes, I know Brad Marchall's work, tryin to comprehend it. I don't oppose O2-/H2O2 theory, on the contrary. Redox state is crucial, but ways to this state can differ. For instance, if PUFAs produce excess of NADP+, cell can get it away, that can cause less NADPH, that can cause less GSH and that can elevate H2O2 and make cell insulin resistant and slows down metabolism. Just like FADH2 defficiency. If the pathway by witch NADP+ is disposed (to NAADP?) elevate gluconeogenesys, then it is even worse. I don't know if it is viable hypothesys, but widening of focus could help, cell is really very complex.
Jaromir
If MTG3Pdh is inputting electrons via Sdh in a similar part of the ETC to EFTdh, and that Sdh input is lowered or inhibited thus lowering the local Redox state would it facilitate input at EFTdh? Making it easier for electrons to fall 'downhill'. Leading to something like the opposite of what is happening with the individual in the paper I linked ie input derived from beta-ox could increase rather than be blocked.
You might have mentioned something along these lines before. So much blog to search!
Post a Comment