This follow on study looking at fructose in people with type 2 diabetes was a bit of a disappointment.
TLDR: Fructose works pretty well in four out of five people with diabetes just as it does in "normal" people with a relatively poor OGTT result.
This is what the title of the study is describing:

It's clear that adding 7.5g of fructose to a 75g OGTT load improved both glucose and insulin curves. There is nothing exciting about this. What I thought might be interesting was that, in a study of five subjects, two of them were outliers of sorts.
I'd hoped these outliers might give more insight in to the state of ROS signalling within hepatocytes of people with severe diabetes. Not really, much of the data you need is not in the paper. For completeness here are the anomalous results:
Subject 2 failed to drop their systemic glucose in response to added fructose, like this

Not unsurprisingly they also failed to drop their insulin level, clearly insulin level follows exposure of the pancreas to systemic glucose, which didn't change, so neither did insulin.
Can we guess what might have been happening in hepatocytes to nullify the "beneficial" effects of fructose? Let's assume that fructose is being absorbed, entering hepatocytes and generating ROS via a NOX enzyme in proportion to the rate of fructose ingress. So the question becomes
"Under what circumstances does a modest increase in ROS fail to activate the insulin cascade?"
We have no idea what the fasting insulin level was, nor the fasting glucose, so this is a little difficult. We are told that subject 2 had an HbA1c of 9.0 or 10.1, one of the highest in the study, which implies the worst average glucose excursion over the last three months or so. We could choose extreme insulin resistance, failing beta cell function or a combination of both to explain this. The fact that they produced a minimal increase in AUC for insulin when presented with 75g of glucose with or without fructose suggests that beta cell function was limited and underlying hyperglycaemia might both be present. I favour this explanation.
Both hyperinsulinaemia -> NOX4 activation via G protein coupled signalling and hyperglycaemia -> NOX2 activation via calmodulin kinase signalling have the potential to spill ROS over from activating via phosphatase inhibition to being inhibitory via insulin receptor substrate inhibition. Adding a small increase in ROS from fructose to a maximally stimulated system may affect both aspects, producing no net change. That's my guess.
The second unusual result was from subject 5. They produced a perfectly reasonable drop in systemic glucose in response to fructose addition but had an unusual increased insulin response despite reduced systemic glucose.
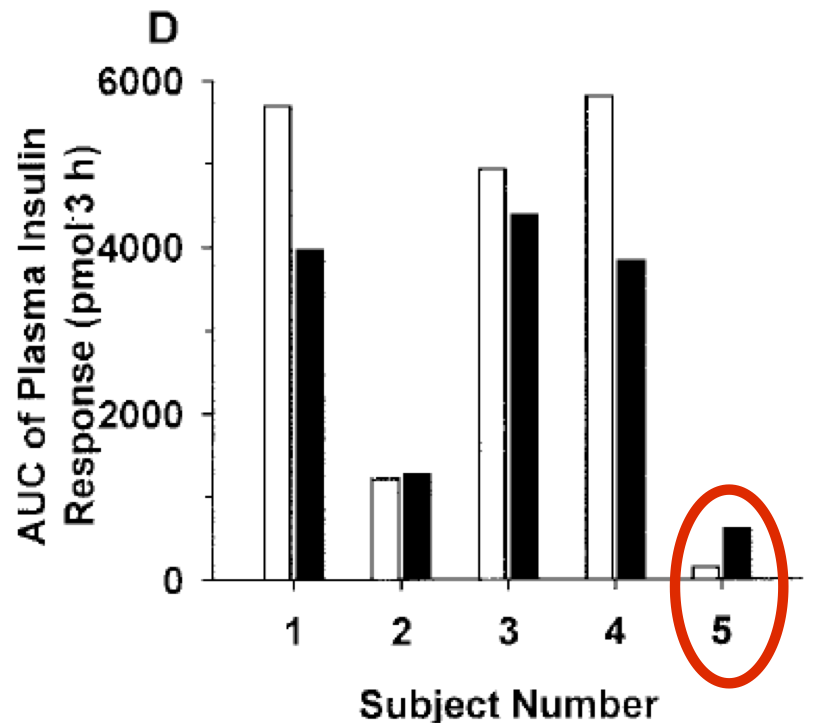
I struggle to explain this "paradoxical" rise in insulin despite a successful fructose induced fall in pancreatic exposure to systemic glucose. You could argue that the increase in insulin came first and this lowered the systemic glucose level but this would make quite an exception and need either a pancreatic response to a very small systemic fructose rise or a pancreatic effect derived from gut hormone signals, none of which were measured and none of which are needed for an explanation of the fructose effect in all other subjects.
If I had to guess I would suggest the fasting insulin level was very high in this person. We know from this 2011 study that OGTTs are reproducible on repeat testing (in "normal" people) over 3 days unless you have to be hyperinsulinaemic in order to be "normal":
"However, our cases of individuals who exhibited hyperinsulinaemia in order to maintain glucose homeostasis, suggest that repeated OGTT’s may not produce a reliable estimation of insulin sensitivity for people with pre-diabetes and diabetes."
Subject 5 was also an individual with very high HbA1c (9.0 or 10.1) suggesting poor control and compatible with high insulin levels. So the insulin anomaly may just be random finding in one person secondary to chronic hyperinsulinaemia... We'll never know.
Overall the fructose effect on hepatic glucose output seems to be genuinely maintained in patients with DMT2 unless they are approaching seriously poor levels of control.
Should individuals with more "mild" diabetes add a little fructose to each bowl of porridge they consume in real life?
Rhetorical question.
Peter

5 comments:
In the Acute Fructose Administration Improves Oral Glucose Tolerance in Adults With Type 2 Diabetes paper - N = only 9.. But they did measure glucose and insulin levels.. but not Fructose levels.
My understanding is that fructose is cleared rapidly??
The question is how much of the insulin level is due to pancreatic response and how much due to the sweetness receptors? Or is the dose high enough that the Sweetness receptors are pegged on? (I would expect other confounders - trygly would be higher in the fructose group - as long as you are drawing blood, why not measure everything?).
.,.
This reminds me of some personal history - back 15 years ago - when I ran OGGT on myself and family members, my BG spiked to almost 140. In the mean time, I greatly reduced carb consumption, gained muscle mass, greatly reduced exposure to PUFA (quitting chicken meat matters here - beef is my health food!). One day I slipped, there was this ginger-bread-sweet - just a little bit - but I ended up eating a huge amount - calculated the sugar exposure was similar to a OGGT test - I was upset with myself - ran a glucose finger test about 1.5 hours later - my level was 94. I was so surprised I ran it again - 92..
So was it because the OGGT was pure glucose and the gingerbread was sucrose? (might be a small effect?), but I think the combination of higher muscle mass and low PUFA intake also matter. What is clear is that T2D can be easily cured with diet and exercise.
What is clear is that if we add the proximal and distal causes of death - T2D is on top. Everyone was so afraid of death by CoVid - yet seem to lack fear of death (and disability) by T2D. Promotion by the press makes a big difference.
Peter, is this effect also dependent on SCD1 and oleic acid? It looks like liver glycogen content is.
Stearoyl-CoA Desaturase 1 Gene Expression Is Necessary for
Fructose-mediated Induction of Lipogenic Gene Expression by
Sterol Regulatory Element-binding Protein-1c-dependent and
-independent Mechanisms*
https://pubmed.ncbi.nlm.nih.gov/15066988/
Jaromir
Hi Jaromir,
I wouldn't be in any way surprised. After all, even before eukaryotes evolved, bacteria/archaea (judging by modern bacterial ROS signalling) have been using ROS to control metabolism. I can't see how manipulating metabolism by altering ROS flux wouldn't adjust every aspect of the follow on metabolism, to achieve the effect of that alteration which is signalled by those extra ROS.
The problems come when inputs are altered to adjust the ROS signal but the end result of that change, in terms of either "aesthetics" or "medicine", don't please the affected individual, be that as obesity or specific pathology.
To look at every individual enzymic aspect of the effector process and to expect to over ride 4.5 billion years of underlying ROS directed evolution carries the risk of becoming a drug developer, trying to "cure" obesity or metabolic syndrome when one should be looking to normalise the ROS signal.
Everything after the ROS signal is metabolic "fluff".
Peter
Fructose, shmuctose. Here's the answer: https://www.youtube.com/watch?v=mGKJX-J4nz4
She seems such a cheerful person. One hopes she doesn't get necrotising fungal vaginitis/urethritis...
Peter
Post a Comment