I have a preference for median lifespan when thinking about the potential effect of an intervention on a population because it is looking at the population as a whole, rather than a few (quirky?) isolated individuals who make up the maximum longevity crowd. Or should I say the maximum longevity chosen few? I guess it confirms my biases too. Which I like.
Let's recap the Jim Johnson's lab findings about the Surwit diet effects described here.
I have removed the lowest insulin gene group data and just included those with a normal insulin phenotype. A number metabolic snapshots were taken at around the 80 weeks of age mark. Surwit fed animals were heavier on the scales and fatter by DEXA compared to chow fed:

No one should be surprised that they were also hyperglycaemic and hyperinsulinaemic, ie insulin resistant:
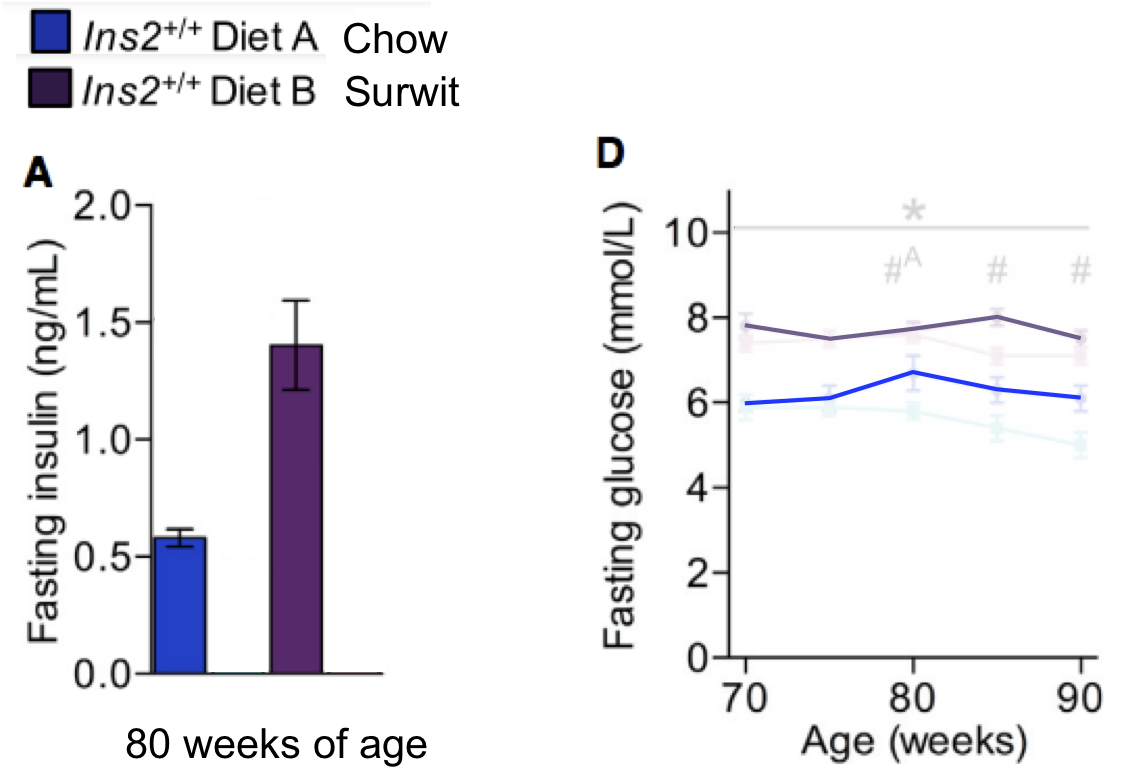
What should be extremely surprising is that these Surwit fed mice while being obese, hyperinsulinaemic, hyperglycaemic and insulin resistant, lived over 100 days longer than their slim chow fed relatives. The data are in here:

which I can simplify down with some very crude curve fitting by eye in Powerpoint to give this:

Interesting.
Now let's look at some rats fed an obesogenic diet based on lard. There are some good data in here although it does not give us the linoleic acid content of the lard:
An isocaloric moderately high-fat diet extends lifespan in male rats and Drosophila
An isocaloric moderately high-fat diet extends lifespan in male rats and Drosophila
Oh, and their data presentation is not great.
Here we have the weights. Colour scheme is different to the first study

You can't tell if the weight loss toward the end was from the surviving rats eating less or that the fattest rats died earliest. Probably a bit of both. But the lard fed rats were fat.
They were hyperinsulinamic

and they were hyperglycamic.

and obviously they too were insulin resistant.
But this time the longevity curves are reversed and the fat rats die younger than the slim control fed rats, by about 100 days:

So we have the mice in Jim Johnson's lab and the rats in the lab in Harbin, China. Both have comparable levels of obesity and insulin resistance. Both are oxidising FFAs when insulin should be suppressing FFA availability in peripheral tissues. In both cases excess energy is being supplied from fatty acids so there is an absolutely normal physiological reduction/rejection of some of the calories which are being taken up by cells using insulin facilitation.
This normal physiological response is mediated by reverse electron transfer through complex I acting to inhibit insulin signalling at the insulin receptor/substrate level.
In animals made obese using fully hydrogenated coconut oil in a Surwit diet the mitochondria are normal and mitochondrial/cytoplasmic membranes lipids are low in linoleic acid as you would expect from 1-2% LA in the diet. So high-physiological ROS from oxidising fatty acids will inhibit the insulin cascade, as they must, but in the process will only encounter "physiological" levels of linoleic acid and only generate "physiological" levels of 4-HNE, 13-HODE, 9-HODE etc. These lipoxides, like superoxide, are normal signalling molecules. A low level of generation is normal and generally beneficial. Probably essential.
Generating ROS via RET in the ETC is pro-survival and pro-longevity. See here, discussion on another day.
The obesity induced by increased dietary linoleic acid is different. Here the adipocytes are large because they fail to limit their insulin cascade adequately. Under these conditions adipose tissues will store an excess of lipid from all sources without insulin being elevated, indeed in the earliest stages insulin signalling will have been enhanced and IR subnormal. The core initiating problem being that linoleic acid is present in levels which generate too small an ROS signal, so fail to limit caloric ingress and storage. Also there is good evidence that linoleic acid is preferentially oxidiseg compared to saturated fats.
However excess basal lipolysis secondary to adipocyte size will release all species of FFAs, arguably with some favourites, the problem now is that adipocytes *ignore* insulin. It doesn't matter how insulin sensitised you might have been via LA in order to become obese. If basal lipolysis is up, it's up. And insulin no longer matters. The exact mix of FFAs delivered to the periphery (especially muscle) becomes less important. All cells oxidising any type of fat generate more ROS than cells oxidising glucose.
One of the secondary problems with high linoleic acid diets is that, separate from their obesogenic effects in mitochondria, the LA molecules are also present at increased levels in tissue lipids.
The problems really present when ROS are being generated on a background of an intake of (in this human observational study) of 17% of calories as linoleic acid. Apart from making you obese this provides an environment where ROS which *should* meet a "normal" level of LA (ie derived from 2% in the diet) are actually encountering an environment derived from 17% LA in the diet. Generating *physiological* levels of 4-HNE etc is out of the question when all the ROS can "see" is freely available double bonds to interact with. Lipids are converted to lipoxides in supraphysiological quantities, cellular damage ensues and this chain of redox damage manifests as what we describe as pathology.
That's what I think is happening.
I think I've said enough for one post. I'll have to write a separate post about RET and Superfly,

the fly featured in the paper nominated for the best graphical abstract of all time, ever. By me anyway.
And about ROS and mitochondrial biogenesis.
Peter

28 comments:
Peter "The obesity induced by increased dietary linoleic acid is different."
That's one of yer classic sentences right there. Perhaps it deserves to be formatted in some special font?
I felt the post had too many italics and asterisks for comfort as it stands! 😂😂😂
P
"These lipoxides, like superoxide, are normal signalling molecules. A low level of generation is normal and generally beneficial. Probably essential."
Yes! Yes!
Superoxide regulates NADH/NAD+ by controlling heat production and oxygen supply from hemoglobin/myoglobin.
Dang, Peter. Nice one
Walter Willett demonized a poor scientist who pointed out that overweight could be healthy.
You show here how that could be true.
We seem to have idealized a suboptimal level of body fat.
Maybe our grandmothers were right.
"Put some meat on those bones!"
As they stuffed us.
I don't know guys, Layne says human outcome studies trump everything - you know, those human outcome studies where things are totally super well controlled over a person's lifespan!
raphi, LMAO,
I know a clown when I see one.
HT @exfatloss
https://twitter.com/exfatloss/status/1714026451731972155/photo/1
Oddly enough Layne is sightly (and accidentally) correct! Eating an obesogenic diet to a normal bodyweight *extends* lifespan, both studies below are based on lard. Calorie restriction doesn't supply the sorts of levels of energy input which will drive RET (or NOX4) to generate the toxic aldehydes which shorten lifespan. The problem is that being hungry for the whole of your life is not much fun! But pathology requires an excess of ROS, physiological ROS won't do it.
https://pubmed.ncbi.nlm.nih.gov/33440166/ Caution, BS title!
https://pubmed.ncbi.nlm.nih.gov/25313149/
I have to say it has occurred to me that a Gymrat might tolerate the obesogenic effects of PUFA if they are abrogating insulin signalling in favour of AMPK signalling for caloric uptake, so lessening the obesogenic effect of over sensitivity to insulin under PUFA.
How does anyone have this sort of a conversation with someone whose health advice is "don't gain fat". Still giggling. But I don't try.
Peter
You need to have glutathione for long lifespan. This is the difference between good and bad fats. And good ROS can trigger synthesis of GSH.
Jaromir
https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=effect+of+hydrogen+peroxide+administration+on+lifespan+musca&btnG=#d=gs_qabs&t=1697715338771&u=%23p%3Dh3a9JeagxBwJ
https://www.mdpi.com/2072-6643/14/5/1114
One more paper, ROS for GSH synthesis, extracellular catalase for GDH depletion.
https://www.tandfonline.com/doi/abs/10.1179/135100003125001260
Cool. Fig 1 of the houseflies fed H2O2 paper is interesting. 4-5mM is approx peak activation of the insulin cascade and 10mM is insulin resistance levels in human adipocytes. Not sure how much H2O2 is needed as a disinfectant to kill bacterial cells, but the ball park figures are fascinatingly. One can speculate about mechansims, which will be multiple. Ultimately, insulin stimulation = short longevity, insulin resistance = increased longevity. Disinfectant levels = death. Also glutathione is fine if it doesn't kill you, which (in flies at least) depends on your mitochondrial/nuclear gene match (unpublished observation, Nick Lane in Transformer).
P
@Peter
This line made my brain hiccup : " insulin stimulation = short longevity, insulin resistance = increased longevity. "
Stimulation is not the opposite of resistance - so it might be better to say:
insulin stimulation = short longevity, insulin sensitivity = short longevity.
Or better yet - the insulin effect is the product of stimulation and sensitivity. ( which implies that we can't look at insulin levels in isolation) and the insulin effect shortens life.
Another way to think of it is there is a cost of the insulin effect. Seems that evolution would happen only if it was trading off against a larger cost? Long term cold survival?
On the other hand, I can see that the evolution of insulin resistance would extends life..
My hunch is the ROS signal is more primitive than insulin itself?
So adding the concept of insulin mimics to this again raises the idea that some of these mimics might be a sort of prototype of the insulin system. The way I'm thinking about this is that to understand biology, it is probably a good idea to try to understand the history of evolution. For instance, lots of enzymes appear to start off as a metal ion that later gets incorporated into a protein that provides feedback loop controls.
Anyway - a very complex system - wonder if we could train a LLM to help make sense of it?
,.,.
I've had a long interest in mitochondrial/nuclear gene match - (hybrid cats often have problems I think due to this mismatch). Could be that an improved definition of what makes a separate species is this match-up between mDNA and nDNA. Probably understanding the details of this match-up would unlock other understandings of how the MT machine works.
,.,.,
RE:"The obesity induced by increased dietary linoleic acid is different."
A title for a future post?
I would say H2O2 function on cellular level as insulin on whole organism level. It suppress lipolysis of fat droplets and let glucose in. Peroxide and NO shows other cells that there is a problem to solve, lack of oxygen, if not solved NO suppress oxygenation and elevates fermentation to correct NAD+/NADH ratio or activate HIF1. The same can be activated by poison like LPS or HPODE, product of LA.
ROS elevate catalase, but only H2O2 is reduced by catalase. So fats peroxides like HPODE elevates catalase without beeng disposed by it. Regulation that works with H2O2 don't work with HPODE.
https://pubmed.ncbi.nlm.nih.gov/10946007/
And what happened if NOX2 is knocked down in mice. Unfortunately catalase was not measured. More and more I think elevated extracellular catalase caused by ROS modulates insulin sensivity.
https://pubmed.ncbi.nlm.nih.gov/23957783/
After reading your "insulin mimesis" series to this post, i wonder...
The story as i have understood is: agents that prioritize the liver or get directed to the liver first (alkohol, fructose, mct, etc) have in small amounts a slimming effect on adipose because they augment insulin signaling at the liver making the liver deal with the substrates (and store them).
In high amounts the same agents will cause IR at the liver, causing the substrates to be exposed to adipose which will have a fattening effect.
In a previous series of yours discussing alcohol you have mentioned that alcohol has "a slimming effect on adipose while grossly fattening the liver" .. which can be of course seen in every alcoholic.
This sounds at the first glance like a contradiction. On the one hand, high amounts of alcohol should fatten adipose and on the other it should and does fatten the liver.
So what am i missing to resolve that contradiction?
"Also glutathione is fine if it doesn't kill you, which (in flies at least) depends on your mitochondrial/nuclear gene match (unpublished observation, Nick Lane in Transformer)." - @Peter
Have you seen this?
Thanks Bob, missed that. Possibly Nick Lane has not updated his personal publications web page since they published! Can't imagine it's a huge priority. Nice to see it in print.
Ta, Peter
Hi Basti,
There is a reply being written.
Peter
Hi Basti,
I’d just rephrase that line “causing the substrates to be exposed to adipose” to my view that hepatic insulin resistances reduces hepatic insulin extraction so allows more *insulin* to the systemic circulation where it can facilitate fat storage in adipocytes. The same hepatic effect also allows more glucose past the liver to stimulate more insulin secretion, which too will also not be extracted by the liver on first pass.
*High* doses of insulin mimetics generate *high* levels of ROS in the peripheral adipocytes and so cause fatty acid release. These FFAs are mopped up by the liver, stored as trigs and destined for export as VLDLs under fasting.
Of note re Surwit diets, under low exposure of adipocytes to MCT there is marked adipogenesis, similar to low dose ethanol/fructose. Unlike ethanol/fructose I doubt you can ever get the sufficiently high levels of MCT exposure to adipocytes needed to force lipolysis in them (easy in vitro), unlike fructose/ethanol which do this at recreational intakes. *Hepatic* exposure to levels of MCTs high enough to cause insulin resistance levels of ROS is quite easy to achieve.
Under fasting the liver only releases VLDLs because insulin signalling is low. If fasting insulin signalling is too *high* in the liver, via LA/Protons (ie too low a level of ROS cf the correct fasting insulin resistance from saturated fats), then excess calories stored in the liver will stay there. And you get hungry. Plus if insulin mimesis in the liver is ongoing because you never stop drinking alcohol or fructose then equally VLDLs will not be released.
Does that resolve matters?
Peter
I spent several hours looking very carefully at all of the qualifiers I needed in that last reply. Apologies for all of the asterisks.
P
Hi karl,
Big delay, your comment triggered a reply which became sufficiently long to be put out as a post. Just posting now.
Ta
Peter
"*High* doses of insulin mimetics generate *high* levels of ROS in the peripheral adipocytes and so cause fatty acid release."
I don't think this is the case. Yes, high ROS is caused by high insulin, but it triggers HIF-1a and lactate production, ROS then fall to low and it's this state that elevates FFA., and as I know, there is no way out of if. And this is done individually, cell by cell. Only apoptosis could be way out, it's like cancer.
Jaromir
And the cell still grow and make new fat, don't need any insulin nor oxygen, only glucose.
An interesting concept. Just looking at fructose as an insulin mimetic driving NOX mediated ROS, how would antioxidants largely reverse the metabolic syndrome induced? My concept is very simplistic (and may be wrong) and doesn't seem to need HIF as a core explanation. I don't doubt that this is related to the pseudohypoxia concept but that seems like a layer on top of the ROS fundamental signal. I don't doubt that HIF is important in view of the hypoxia event preceding the later rise in oxygen and the Cambrian speciation explosion. Very important, but I struggle with fundamental. I may have to change my mind one day.
Peter
One cannot be sure with anything, insulin not elevate OXPHOS, but likely elevate glycolytic ATP. Nitrite relocates GLUT4 etc.
Fructose elevates glycolytic ATP from glucose by attaching to PKM2 and for lactate to be released by MCT4, HIF1 has to be activated. But this also activates lipogenesis. It looks like insulin function now. So NOX will be activated by FFA waiting for oxidation may be.
Jaromir
https://pearl.plymouth.ac.uk/handle/10026.1/18863
And with HIF I can easily explain even such thing as how fruits suppress HIF by flavonols and make protection against fructose so preserve OXPHOS.
https://mct4health.blogspot.com/2022/12/onion-chocolate-tea-or-wine-flavonols.html
Okay thats again a lot of information for me to process. And yeah i guess it cleared thinks up.
Thanks a lot for your care.
Thanks for the clarification in your first paragraph.
So the nuance is that alcohol/fructose reach the liver as well as peripheral adipose at low AND high doses.
At low doses they will fatten both due to augmented insulin signaling. At high doses though, the lipolysis from the IR adipocytes overwhelms the IR liver.
So the net effect is a fatty liver tendency. No matter the dose. Just more so with high doses.
Is that correct?
Next up is the situation under LA...
Due to its effects it should aggrevate the fatty liver tendency of low doses of fructose/alcohol because it allows more fat storage before resistance sets in.
Equally though, under high doses, it should LESSEN the fatty liver tendency, because this time, the later occurance of resistance in adipocytes should decrease lipolysis.
So based on that reasoning a high LA diet should from the protons perspective be preferred over a low LA diet when you choose to consume high amounts of alcohol/fructose.
Shouldnt it?
The reason the opposite is probably true lies in the other effects LA is said to bring with it. (Turning into OXLAMs and such).
(This is my explanation why high LA alcoholism is worse than low LA alcoholism even though protons would predict the opposite)
Or where is my faulty or (incomplete?) logic applied?
ok, while Protons suggests amelioration of hepatic lipidosis via augmenting adipocyte insulin signalling, this can be over-ridden by gross ROS generation (via NOX) of adipocyte exposure to high ethanol induced ROS -> insulin resistance. Leading to weight loss. Ditto fructose. As you correctly suggest, hepatic lipid peroxides make LA pure badness in the liver where ROS are also high from ethanol/fructose...
Peter
Peter, as I go through papers, I see that "metabolic" insulin resistance can be the lost ability to go from OXPHOS to glucose fermentation immediately. This can cause lack of cytosolic NADH when desperately needed (by NAD+ deficiency?).
You show less insulin is protective. I send paper with NOX2 KO (not NOX4, why?) so less ROS is protective. Mouse is insulin resistant but stay AKT and eNOS sensitive. So there are healthy "metabolical" insulin resistance that can be overloaded to pathological "epigenetical" insulin resistance (or pathological insulin sensitivity?), we have to differentiate them. What is exactly the mechanism if not HIF gene expression change?
Jaromir
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6115650/#:~:text=In%20diabetic%20cells%2C%20increased%20fatty,blunting%20the%20adaptation%20to%20hypoxia.
Post a Comment