Here's the next paper:
I would suggest that the first of many problems with the paper is that it is using ob/ob and db/db mice. I don't know about ob/ob mice but in db/db mice we already know that obesity is only a feature of the genotype when linoleic acid is present in the diet at somewhere above 5% of calories. I discussed Valerie Reeves' PhD here. So the db/db mouse obesity phenoptype is really just a consequence of the poor ROS generation exemplified by linoleic acid. Offsetting this with stearic acid is 100% protective against obesity.
Think about that.
If anyone goes through the paper they will also note that the adipocytes from these mice which exhibit obesity under linoleic acid via low ROS generation are also under extreme oxidative stress, ie high ROS generation, during the process of dying. That's yet another post which will have to wait.
On the plus side this suggests that what we see in db/db mice has the potential to be relevant to what happens in D12942 fed bl/6 mice, which the group didn't use for their electron micrographs here. My bias is that db/db mice are remarkably similar to D12942 fed mice in their phenotype. Others may disagree.
So this is a scanning electron micrograph of the degenerating remains of an adipocyte from a db/db mouse:

The large spherical surface is the dying adipocyte. It still has a surface structure of some sort, which is covered in collagen fibrils. See images in Fig 4 for more beautiful microscopy of this. I would suggest that the collagen has been secreted by the macrophages, as they do,
Production of type VI collagen by human macrophages: a new dimension in macrophage functional heterogeneity
Production of type VI collagen by human macrophages: a new dimension in macrophage functional heterogeneity
to maintain localisation of the large lipid droplet from the dead adipocyte while they deal with it. Okay, okay, here's an histo image from Fig 4, stained for collagen:

and here's a scanning EM of the collagen fibrils on the surface of such an adipocyte, or its remains:

If we go back to a selection of the scanning EM from the top of the post we can see, apparently oozing from the surface, are small lipid droplets (arrows), at least one of which (arrowhead) has been eaten by a macrophage. Each asterisk identifies a macrophage:

We can get an idea of what happens to the lipids in these droplets from here:
Well, well, well. Macrophages release of FFAs from the (remains of) adipocytes, a process which, I very much expect, cannot be suppressed by insulin. Would anyone have any expectations as to whether this process, irrespective of any sort of johnny-come-lately-add-on cytokines, might *require* insulin resistance? Evolution builds on what was there already. With modifications.
Where do these FFAs end up if they can't be constrained within adipocytes, for what ever reason? In part they might, if you eat some carbohydrate, be pushed back in to other adipocytes and trigger even more CLS formation, more lipid release, more... Oh wait, week 16 of D12942 feeding looks like this!

Of course the FFAs also end up, to a very large extent, in the liver when adipose cannot mop them up adequately. From the previous CLS paper again:
"However, under conditions of chronic HF feeding, eAT reduction could contribute to lipid overflow to the liver (Fig. 6), potentially exacerbating hepatic insulin resistance (50) or promoting steatohepatitis."
Here's Fig 6:

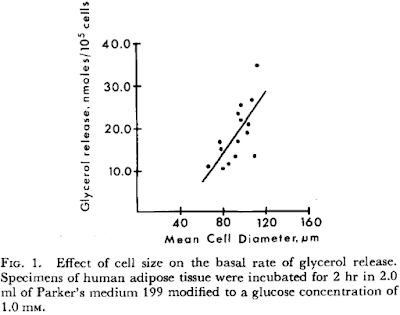
I look at CLS formation as the end stage of a failure of rising basal lipolysis to limit linoleic acid mediated adipocyte distension. It has the same consequences but more so.
Peter