Tucker has a podcast episode in which he chats to John Speakman about obesity. It's one of the more interesting podcasts I've listened to in many years.
Ep. 22: John Speakman—What Causes Obesity?
A very large part of the core discussion is contained within this paper, a massive collaboration, with Speakman as first author:
Total daily energy expenditure has declined over the last 3 decades due to declining basal expenditure not reduced activity expenditureEp. 22: John Speakman—What Causes Obesity?
A very large part of the core discussion is contained within this paper, a massive collaboration, with Speakman as first author:
Basically total daily energy expenditure in the studied populations is down slightly over the last 30 years, despite daily activity energy expenditure going up. This means that basal metabolic rate must have dropped.
Which, of course, begs the question of what might cause basal metabolic rate to fall.
The answer is not obesity.
There are certain groups of people who *do* have a decreased BMR, the most obvious of whom are the post-obese.
The post-obese, like the pre-obese, come with a cluster of abnormalities the two most prominent of which are an enhanced insulin sensitivity and a defect in fat oxidation. And sometimes a depressed metabolic rate, especially BMR.
To me, the enhance insulin sensitivity is causal, the impaired fat oxidation is secondary. The decreased metabolic rate is simply a longer term downstream effect of chronic under supply of calories to metabolism.
Aside: I haven't discussed it yet but, obviously, pathological insulin sensitivity should also show as an exaggerated ability to over-store fat under peak insulin effect. This shows rather nicely under an hyperinsulinaemic euglycaemic clamp in Astup's lab. See top panel of Fig 2. But currently I'm mostly thinking about fasting conditions. End aside.
So. The core feature of pre or post obesity following on from the pathological insulin sensitivity is a decreased ability to oxidise lipid and a facilitated ability to oxidise carbohydrate. The RQ should rise.
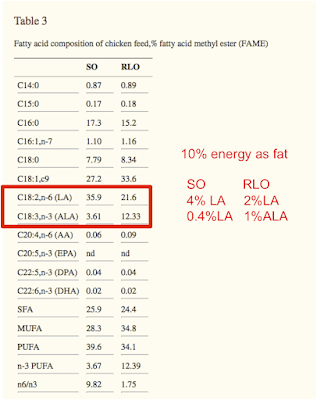
What would happen if you took eight apparently healthy men and fed them, for a week, a complete diet providing 2% PUFA then switched them to a 10% PUFA diet for another week, as a crossover study?
This is the paper, from 1988:
Polyunsaturated:Saturated Ratio of Diet Fat Influences Energy Substrate Utilization in the Human
You can clearly alter the RQ under fasting conditions, on a fixed food quotient diet, simply by altering the dietary fat from 2% of calories as PUFA to 10% PUFA, switching palmitate in or out to balance the PUFA, which was mostly linoleic acid. MUFA were kept constant, as were all other macros.
Polyunsaturated:Saturated Ratio of Diet Fat Influences Energy Substrate Utilization in the Human
You can clearly alter the RQ under fasting conditions, on a fixed food quotient diet, simply by altering the dietary fat from 2% of calories as PUFA to 10% PUFA, switching palmitate in or out to balance the PUFA, which was mostly linoleic acid. MUFA were kept constant, as were all other macros.
Within seven days this happened to the fasting RQ values.
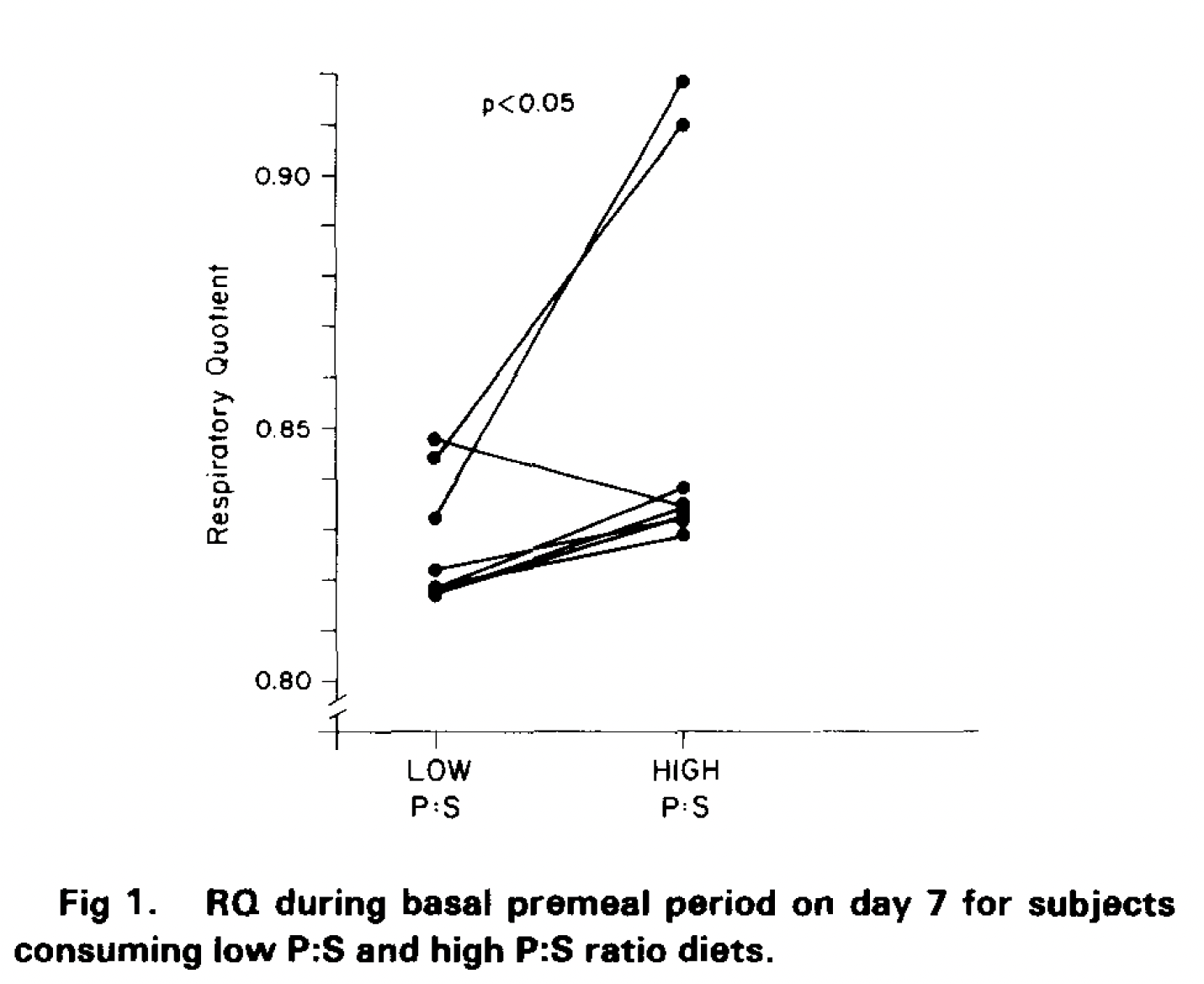
Obviously there are three interesting subjects. One showed a decrease in RQ, suggesting enhanced lipid oxidation under linoleic acid. That's unusual. It is normal for linoleic acid to augment the thermic effect of food because it is preferentially oxidised but that is finished well before an overnight fast is finished. Hard to say what was going on with that subject. It wasn't a hospitalised study but all food was provided by the investigators. File it under odd.
The rise in RQ, signifying a change away from lipid and towards carbohydrate oxidation while fasting, was (pax the exception) ubiquitous across all other subjects, but in two subjects there was such a rise in RQ that the investigators seriously considered that there might be a problem with their measurement system. There wasn't. Their comment:
"Although a fasting RQ of 0.9 is unusual, reanalysis of the calibration parameters of the respiratory gas exchange system obtained prior to tests on these subjects revealed no abnormality in analyzer response. No reason for rejection of these RQ values could be determined."
Clearly 10% of LA in the diet moves almost all subjects towards a "pre-obese" phenotype. In two of the eight this move was dramatic. It seems very, very likely to me that these two individuals are at serious risk of obesity in an omega-6 rich environment. Follow up weights over the years would have been lovely but was not remotely the purpose of the study.
Clearly 10% of LA in the diet moves almost all subjects towards a "pre-obese" phenotype. In two of the eight this move was dramatic. It seems very, very likely to me that these two individuals are at serious risk of obesity in an omega-6 rich environment. Follow up weights over the years would have been lovely but was not remotely the purpose of the study.
You can, within seven days, convert normal people in to pre-obese people, as viewed from metabolic substrate oxidation perspective.
All you have to do is make sure they are eating 10% of their calories from linoleic acid.
Some people will get bitten by this feature of linoleic acid more rapidly than others.
Eventually the whole population will.
Thank your cardiologist.
Peter
Addendum. The world is full of U shaped curves. Adding linoleic acid to the diet causes an initial excess insulin sensitivity. This distends adipocytes. As adipocytes distend they increase their basal lipolysis and release FFAs which cannot be suppressed by insulin. This, at some point, appears to normalise fasting insulin sensitivity at the cost of distended adipocytes, ie obesity, and chronically elevated FFAs. On a starch based diet the high level of post prandial insulin needed to overcome the still (unsupressable) FFA induced insulin resistance at peak absortption will sequester more lipid in to adipocytes, from where they will again leak, via basal lipolysis, leading to frank insulin resistance, hyperinsulinaemia and metabolic syndrome.
Under fasting conditions the pathological insulin sensitivity activates malonyl-CoA formation and the subsequent inhibition of CPT1 mediated entry of fatty acids in to mitochondria. This would, if it occurred in isolation, simply lead to hypometabolism unless enough glucose alone was available to run metabolism. However, it doesn't happen in isolation. It happens combined with obesity, which increases the supply of FFAs irrespective of insulin sensitivity. All that is needed is to elevate FFAs high enough to get adequate substrate in to mitochondria (there is not 100% inhibition of CPT1) and enough lipid derived ROS can then inhibit insulin, reactivate CPT1 and restore metabolism. Hence obese people have high metabolic rates.
The crux comes with conventional dieting. As adipocytes shrink the supply of FFAs from basal lipolysis drops, insulin sensitivity is restored and people get right back to where linoleic acid takes them: obtunded fat oxidation, carbohydrate dependency and hypometabolism. The classical post-diet hungry person.
Why is BMR falling in the developed world despite obesity being rampant? Because everyone is being drugged with linoleic acid to become obese and no one wants to be fat. The more you resist obesity, the more your caloric restriction shows as decreased BMR. The BMR is falling in response to Weight Watchers, Slimming World etc. People are not as fat as linoleic acid "wants" them to be.
Ultimately obesity "fixes" the pathological insulin sensitivity from linoleic acid on both fronts, at the cost of weight gain. But it's not a real fix, it's a sticking plaster and we call it metabolic syndrome.
End.