Time to get back to the blog. We had a great summer and life has just kept on being more interesting than blogging. There are about 20 comments I need to read and approve which I'll do my best to get around to, but I thought it was time to hit the keyboard after the summer holidays.
I thought I would just post briefly on the struggles of trying to work out exactly what a given paper is describing in dietary research. I did set out a post a few weeks ago, being rather derogatory, about this paper on ALS. Here is the preamble:
I feel I should like the paper. Really. What with all this iced water being poured over people's heads in the name of ALS research etc. But it's hard.
OK. We're looking at a ketogenic diet for mice endowed with an engineered model of ALS which is quite similar to one of the familial forms of human ALS.
Being me, I go to the methods section first, to see what they fed the poor mice on. From the philosophical point of view I expect the methods section of a paper to allow me to duplicate a given research protocol. All I am told in this case is that the ketogenic diet is 60% fat, 20% carbs and 20% protein and that it was made by Research Diet, Inc. New Brunswick, NJ. That's it. Now, until RD are bought out by some other multinational company, they have a website and this tells me that they supply only one ketogenic diet, D12336, which is 11% protein 89% fat and zero carbohydrate, pretty much what you need to get a rodent in to mild ketosis. So this research group are using a custom diet, what goes in to it is anyone's guess.
My guess is medium chain triglycerides. I don't think you can get a mouse in to ketosis with protein at 20% of calories and carbohydrate at 20%. You'd have trouble getting a human in to ketosis with this, unless you used MCTs.
This is important because I'm interested in teasing out whether there is any point in the enormous effort and endless tedium of eating a low carbohydrate driven ketogenic diet with thyroid deficiency, lethargy, brain fog, glucose deficiency and auto immune disease predisposition as routine sequelae, not to mention the constipation and halitosis (is this me?), when merely popping down to my local Caribbean corner store for a bottle of coconut oil might do the job equally well.
What goes in to the diet matters. Coconut oil is not safflower oil, is not butter. What goes in to the methods matters. Research must be replicable.
End of preamble. I wasn't best impressed.
Before I go on to think about ALS and what help medium chain triglycerides may or may not provide in another post, I thought I would just like to revisit the lethal effects of a VLC keogenic diet paper on the outcome of induced ischaemia and reperfusion of the myocardium in some hapless rats.
It is fairly clear that using "vegetable shortening" as your primary ketogenic source of calories is likely to destroy your myocardium if you have an ischaemic episode. Your first heart attack might be your last. It took an email to Research Diets to get the information about the probable trans fat content of their diet and confirmed to me that the research group had written a methods section which put their paper, and probably the researchers, in to the garbage category.
The same appears to apply to the flip side. I had wondered what a non vegetable shortening based ketogenic diet might do under the same circumstances. Well I'd missed the study, which fully reinforces my pro-LC confirmation bias. Ketogenic diets are the bees-knees for surviving a period of cardiac ischaemia, Crisco excepted.
So, what does the miracle diet for surviving your next coronary look like? I don't know. You don't know. You can read the full text. You still won't know.
The diet is 60% protein by calories! And 10% carbohydrate. The remaining 30% is "oil". Now you know as much as in the paper. Can you replicate the study, based on the methods? No.
BTW: They didn't even check ketone levels! I think we have to assume MCTs again and assume some degree of ketosis.
Crap.
The scrutineers also need to be up against the wall come the revolution.
For all three papers. Crap. Even though I like the results.
Hiya all!
Peter
Sunday, September 21, 2014
Friday, July 11, 2014
Neuron fuel and function
In the comments following a previous post Dustin linked to this rather lovely paper from the early 1970s, back when I was still at school and marathon racing my kayak.
This is one of the nicest figures:

The lower section is the interesting part. CJ had not eaten for 50 days and had a (very) fasting blood glucose of 4.0mmol/l when he received around 0.1 IU/kg of insulin by intravenous injection. Please don't try this at home. You can see that a) he is still insulin sensitive and b) his blood glucose bottomed out at around 0.5mmol/l by 60 minutes. Throughout this period he was asymptomatic. No hypo.
CJ was not running his brain on glucose. The upper section shows a rapid and sustain increase in B-OHB extraction (the dark hatching between the arterial line and jugular bulb line for B-OHB) by his brain through this period of time. Ketones, at levels in excess of 11.0mmol/l, can sustain apparently normal brain function. Given an alternative fuel source this would seem to put the level of blood glucose needed for normal neural function at some (non determined) value of less than 0.5mmol/l.
Drenick et al did not look at fatty acid extraction by the brain. That's a pity, but understandable. No one expects the brain to metabolise palmitic acid. Well, perhaps we should say that neurons should not metabolise palmitic acid. Astrocytes do. Astrocytes are ketogenic and are in a perfect position to supply ketone bodies to neurons using the monocarboxylate transporters ubiquitous on them. I hope to come back to this by the end of the post.
Of course, a 50 day starvation period is not exactly the normal human predicament and we could argue that the normal human brain is glucose dependent. This too, I think may be a very debatable point.
Work has been done with humans under hypoglycaemia with brain function supported by either lactate or pyruvate by intravenous infusion. They're fairly effective, not perfect, but there are limits in how far you can push the metabolism of human volunteers.
Rats are not so fortunate.
If we go to Figure 1 from the rather nice paper emailed to me by Edward

we can see that, under the influence of a massive 20 IU/kg of insulin, there is an almost complete loss of plasma glucose and a slightly more complete loss of brain response to limb stimulus in the insulin-only, profoundly hypoglycaemic group, top row of section A. This has occurred by two and a half hours. The next row has had the hypoglycaemia corrected with glucose (i.e. it's essentially a control group) and has a normal response to stimulus at 4 hours. The lower row shows the effect of lactate in supporting brain function during four hours of persistent, profound, uncorrected hypoglycaemia. You have to note that progress from left to right is time in milliseconds after stimulus and that there is a clear cut delay of about 10 ms in the response time under pure lactate compared to under glucose. This is reiterated in section D, where the response can be seen to be delayed and blunted when compared to the glucose supported data of curve C.
This has led the authors to speculate that, heresy of heresies, there may actually be an absolute need for some glucose by the brain! Strange I know, but... They're not sure of this, just speculating. There are other potential explanations.
Now, most people do not walk around with a blood lactate of 9.0mmol/l. Perhaps most people really do run their brain on glucose?
This seems very unlikely. Or, rather, it seems very unlikely that the neurons in the brain run on glucose. Astrocytes certainly do. But one of the main functions of astrocytes appears to be to manufacture lactate from glucose (directly or from stored glycogen) and deliver it to neurons as a one step conversion fuel giving pyruvate, which can enter the TCA as acetyl-CoA without any messy glycolysis. There is an awful lot of information in this paper.
I put up this nice illustration previously, in the Protons thread:

Neurons are spared glucose. Why?
Mitochondrial glycerol-3-phosphate dehydrogenase. Glucose, during glycolysis, is quite able to input to the electron transport chain through an FADH2 based input at mtG3Pdh, which can reduce the CoQ couple and set the ETC up for reverse electron flow through complex I, with the generation of superoxide as this occurs. Modest superoxide is a Good Thing, especially if you want to signal for mitochondrial biogenesis or cell division. Excess superoxide is a potent signal for apoptosis. Apoptosis is verboten for CNS neurons because any information stored in their synaptic connections will be lost along with the cell. Replacing the cells will hardly replace the memories and appears equally forbidden. There is a need for immortality without reproduction in neurons, for their survival in excess of the lifespan of an organism needing a functional memory in a learning brain.
Feeding lactate through pyruvate to acetyl-CoA does not drive CoQ pool reduction "ahead" of the throughput of electrons coming in from complex I. Neurons do not want to generate insulin resistance. Avoiding glycolysis looks (to me) to be the way they do this. Generating hyperglycaemia looks like a way to overcome the normal lactate shuttle and of forcing glucose directly in to neurons. Enough apoptosis and eventually neural loss just might show as memory loss.
Hyperglycaemia and Alzheimer's...
Generating large amounts of superoxide in astrocytes during glycolysis is not damaging to the neurons supported by the derived lactate. Astrocytes are certainly replaceable, although there seems to be some debate about cell division vs stem cell recruitment. Astrocytes are also able to divide unreasonably rapidly and form various grades of brain tumour. They are common and frequently aggressive. Neuron derived tumours are much rarer and are usually derived from embryonic cells giving medullablastomas rather than being derived from mature neurons. That seems to fit the metabolic arrangements in the brain rather neatly.
This takes us back, eventually, to palmitate as a ketogenic energy supply to the brain via astrocytes. Again, an FADH2 input through electron transporting flavoprotein dehydrogenase can couple with hyperglycaemia to generate reverse electron flow through complex I giving excess superoxide generation. I consider this to be why free fatty acids are excluded from neurons. It's not that FFAs generate excess superoxide per se, they don't. But combined with hyperglycaemia they certainly do, especially palmitate and the longer of the saturated fatty acid series. You really don't want this happening in a cell whose remit is immortality.
Ketones and lactate do not drive reverse electron flow through complex I. Glucose can. Palmitate certainly can. What you want from a metabolic fuel depends on the remit of your cell types. Neurons within the brain preserve information by their continued existence. This is best done by burning lactate or ketones. NOT glucose and, of course, not FFAs. Anyone who claims that glucose is the preferred metabolic fuel of the brain has not though about what a neuron has to do and what an astrocyte actually does do. Or much about the electron transport chain.
Peter
This is one of the nicest figures:
The lower section is the interesting part. CJ had not eaten for 50 days and had a (very) fasting blood glucose of 4.0mmol/l when he received around 0.1 IU/kg of insulin by intravenous injection. Please don't try this at home. You can see that a) he is still insulin sensitive and b) his blood glucose bottomed out at around 0.5mmol/l by 60 minutes. Throughout this period he was asymptomatic. No hypo.
CJ was not running his brain on glucose. The upper section shows a rapid and sustain increase in B-OHB extraction (the dark hatching between the arterial line and jugular bulb line for B-OHB) by his brain through this period of time. Ketones, at levels in excess of 11.0mmol/l, can sustain apparently normal brain function. Given an alternative fuel source this would seem to put the level of blood glucose needed for normal neural function at some (non determined) value of less than 0.5mmol/l.
Drenick et al did not look at fatty acid extraction by the brain. That's a pity, but understandable. No one expects the brain to metabolise palmitic acid. Well, perhaps we should say that neurons should not metabolise palmitic acid. Astrocytes do. Astrocytes are ketogenic and are in a perfect position to supply ketone bodies to neurons using the monocarboxylate transporters ubiquitous on them. I hope to come back to this by the end of the post.
Of course, a 50 day starvation period is not exactly the normal human predicament and we could argue that the normal human brain is glucose dependent. This too, I think may be a very debatable point.
Work has been done with humans under hypoglycaemia with brain function supported by either lactate or pyruvate by intravenous infusion. They're fairly effective, not perfect, but there are limits in how far you can push the metabolism of human volunteers.
Rats are not so fortunate.
If we go to Figure 1 from the rather nice paper emailed to me by Edward
we can see that, under the influence of a massive 20 IU/kg of insulin, there is an almost complete loss of plasma glucose and a slightly more complete loss of brain response to limb stimulus in the insulin-only, profoundly hypoglycaemic group, top row of section A. This has occurred by two and a half hours. The next row has had the hypoglycaemia corrected with glucose (i.e. it's essentially a control group) and has a normal response to stimulus at 4 hours. The lower row shows the effect of lactate in supporting brain function during four hours of persistent, profound, uncorrected hypoglycaemia. You have to note that progress from left to right is time in milliseconds after stimulus and that there is a clear cut delay of about 10 ms in the response time under pure lactate compared to under glucose. This is reiterated in section D, where the response can be seen to be delayed and blunted when compared to the glucose supported data of curve C.
This has led the authors to speculate that, heresy of heresies, there may actually be an absolute need for some glucose by the brain! Strange I know, but... They're not sure of this, just speculating. There are other potential explanations.
Now, most people do not walk around with a blood lactate of 9.0mmol/l. Perhaps most people really do run their brain on glucose?
This seems very unlikely. Or, rather, it seems very unlikely that the neurons in the brain run on glucose. Astrocytes certainly do. But one of the main functions of astrocytes appears to be to manufacture lactate from glucose (directly or from stored glycogen) and deliver it to neurons as a one step conversion fuel giving pyruvate, which can enter the TCA as acetyl-CoA without any messy glycolysis. There is an awful lot of information in this paper.
I put up this nice illustration previously, in the Protons thread:
Neurons are spared glucose. Why?
Mitochondrial glycerol-3-phosphate dehydrogenase. Glucose, during glycolysis, is quite able to input to the electron transport chain through an FADH2 based input at mtG3Pdh, which can reduce the CoQ couple and set the ETC up for reverse electron flow through complex I, with the generation of superoxide as this occurs. Modest superoxide is a Good Thing, especially if you want to signal for mitochondrial biogenesis or cell division. Excess superoxide is a potent signal for apoptosis. Apoptosis is verboten for CNS neurons because any information stored in their synaptic connections will be lost along with the cell. Replacing the cells will hardly replace the memories and appears equally forbidden. There is a need for immortality without reproduction in neurons, for their survival in excess of the lifespan of an organism needing a functional memory in a learning brain.
Feeding lactate through pyruvate to acetyl-CoA does not drive CoQ pool reduction "ahead" of the throughput of electrons coming in from complex I. Neurons do not want to generate insulin resistance. Avoiding glycolysis looks (to me) to be the way they do this. Generating hyperglycaemia looks like a way to overcome the normal lactate shuttle and of forcing glucose directly in to neurons. Enough apoptosis and eventually neural loss just might show as memory loss.
Hyperglycaemia and Alzheimer's...
Generating large amounts of superoxide in astrocytes during glycolysis is not damaging to the neurons supported by the derived lactate. Astrocytes are certainly replaceable, although there seems to be some debate about cell division vs stem cell recruitment. Astrocytes are also able to divide unreasonably rapidly and form various grades of brain tumour. They are common and frequently aggressive. Neuron derived tumours are much rarer and are usually derived from embryonic cells giving medullablastomas rather than being derived from mature neurons. That seems to fit the metabolic arrangements in the brain rather neatly.
This takes us back, eventually, to palmitate as a ketogenic energy supply to the brain via astrocytes. Again, an FADH2 input through electron transporting flavoprotein dehydrogenase can couple with hyperglycaemia to generate reverse electron flow through complex I giving excess superoxide generation. I consider this to be why free fatty acids are excluded from neurons. It's not that FFAs generate excess superoxide per se, they don't. But combined with hyperglycaemia they certainly do, especially palmitate and the longer of the saturated fatty acid series. You really don't want this happening in a cell whose remit is immortality.
Ketones and lactate do not drive reverse electron flow through complex I. Glucose can. Palmitate certainly can. What you want from a metabolic fuel depends on the remit of your cell types. Neurons within the brain preserve information by their continued existence. This is best done by burning lactate or ketones. NOT glucose and, of course, not FFAs. Anyone who claims that glucose is the preferred metabolic fuel of the brain has not though about what a neuron has to do and what an astrocyte actually does do. Or much about the electron transport chain.
Peter
Wednesday, June 25, 2014
Cardiac ischaemia and low carbohydrate diets
There are researchers within the worlds of both nutrition and cardiology who appear to be very determined to supply a message that failing to eat adequate carbohydrate is a very dangerous choice. They occasionally produce little gems of research.
This paper, which has been sitting on my hard drive for a few years, surfaced on Facebook recently. I gather it has been cited by someone who’s writing requires more ondansetron to read than I currently possess. I’ll just assume it was some sort of “eat LC and the first smidgin of myocardial hypoxia will finish you off” warning, but I’m just guessing and I have every intention of keeping it that way.
The paper itself is very convincing, well written and the protocol extensively justified. The core findings are that a LC diet impairs insulin signalling, depletes myocardial glycogen and results in massive necrosis during reperfusion after a period of myocardial hypoxia. The basic idea is that the lack of glycogen limits substrate for anaerobic glycolysis and failed insulin signalling both impairs glucose delivery from the perfusate and also fails to deliver a number of highly beneficial insulin effects which are independent of GLUT4 translocation.
This is the fate of the LC myocardium. As one of my co workers might say: Dig the hole, choose the coffin.

Obviously, for a LC eater, this is disturbing. The queue for McDougall-ism is over there.
The first thing which I find slightly disturbing is that, in a trial of the Atkins Diet™ (always mention by name), the rats ate more and were significantly heavier than the control rats, within two weeks. I’m not totally certain if I remember correctly, but I thought that the Atkins Diet was used for weight LOSS, not weight gain. Perhaps the authors might have been a little disturbed by this finding too, but apparently it doesn’t need mention. Hmmm.
The Atkins™ like diet is TestDiet 5TSY, no longer manufactured. Table I is abstracted by the authors from the full formula provided by Purina and supplies information on a need-to-know basis.
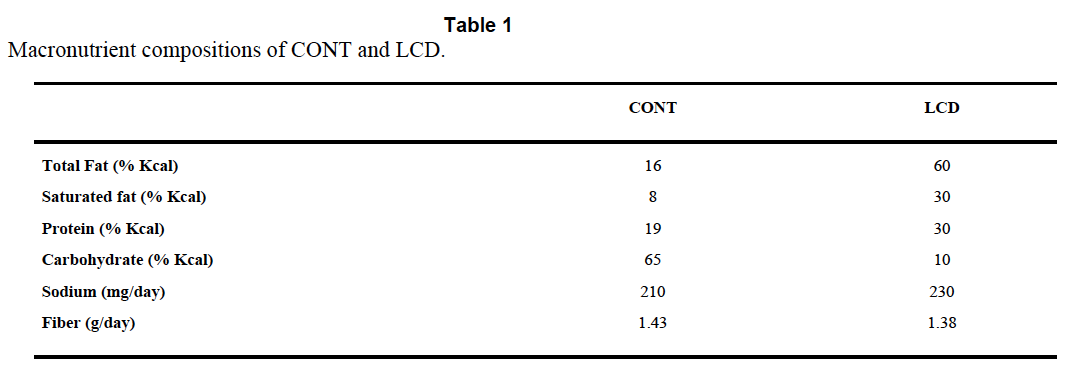
So we know total fat, saturated fat and that “The diets have the same concentrations of essential fatty acids”, i.e. we don’t quite know what the diets were made of. But Purina still will email you a pdf (very promptly) and that gives you this:

There is no mention of Crisco™ by name and no information about the trans fat content, but can you guess how much "Vegetable shortening" is in the control diet? Oh, you guessed!
I know it seems stupid to say this, but if you want to lose weight and/or survive a heart attack, for goodness sake do not choose Crisco or its equivalent as 20% of your calorie intake.
Now, this study does not tell us that a low carbohydrate diet is is good or bad for surviving a heart attack, it's simply not possible to pull that information out due to the lack of control of the variables in the diets. One can only wonder whether the formula of the Atkins Rodent Diet was specifically developed to cause metabolic problems or that somewhere along the line the original Atkins diet suggested a generous consumption of trans fat based vegetable shortening. Maybe I missed this.
I guess you could leave it there and say don't eat vegetable shortening, but there are a whole stack of follow on ideas to this study. There is MASSIVE cardiac necrosis in the LC group of rats. If it is the trans fats, how do they cause this?
One very interesting aspect of the study is the pre-ischaemia depletion of glycogen. These rats are not in ketosis. The doubling of ketone levels from 0.3mmol/l to 0.6mmol/l may well be statistically significant, but is not biologically significant. To go back, yet again, to Veech et al, we need around 5.0mmol/l of mixed ketone bodies to completely replace the insulin signalling system. You can't sidestep insulin resistance with 0.6mmol/l B-OHB. You simply cannot get rats in to a functional level of ketosis with protein at 30% of calories and carbohydrate at 12%. You need carbs near zero and protein limited to < 10% of calories to have rats in nutritional ketosis. Some papers limit protein to < 5% of calories, with minimal carbohydrate.
A period of starvation for rats on the control chow would have tested the hypothesis that it was a lack of glycogen which damaged the myocardium under hypoxia.
My own idea is that the glycogen depletion might be a surrogate for insulin resistance rather than carbohydrate restriction. We have long known that trans fatty acids from partially hydrogenated vegetable oils induce insulin resistance (though not in every study!).
So the next thought is: What sort of insulin resistance?
Are we thinking about an excess of superoxide from complex I, to imitate post prandial hyper caloric insulin resistance? Or are we thinking about the insulin resistance of fasting, when uncoupling results in blunted insulin signalling combined with high oxygen consumption and limited ATP generation?
I like the second idea when applied to this study.
If trans fatty acids, which are structurally similar but not quite identical to saturated fatty acids, allow persistent low level uncoupling outside of the physiological role of the normally structured FFAs it is interesting to speculate that they may continue to allow uncoupling when uncoupling should be utterly and totally banned.
The consequences would be decreased ATP production per unit oxygen consumed. A bit like the findings in Table 3 of this paper. The last column gives you the ATP generated per unit O2 consumed. This would be a disaster under hypoxic conditions. The IF line uses industrial trans fats. The soleus muscle is the mitochondrial dependent one cf the tibialis muscle..

ASIDE: It's worth noting that cytoplasmic ATP is a marked inhibitor of uncoupling but that this is easily overcome by adequate levels of mitochondrial ATP binding within the UCP pore from the end which does not produce a conformational change. Figure 7 gives the details but the main text is really clever stuff (without an agenda, as far as I can see). I'll blog about this paper sometime.

Under this control system the occurrence of falling mitochondrial ATP levels should allow cytoplasmic ATP to immediately shut down the normal uncoupling associated with ketogenic eating and maximise coupled ATP production per unit oxygen when this is needed. The interesting question is whether the uncoupling suspected of transfats persists under low mitochondrial ATP levels. END ASIDE.
You could speculate for hours about what trans fats may or may not do.
This is a very fertile area for idea generation. Ultimately, we don't know what a LC diet based on real Food would do to ischaemic damage in the heart. Maybe it will be as bad as a trans fat based LC (weight gain inducing) diet, maybe less severe.
We are never going to find out what Food does by using "AIN-93G Atkins/Rodent 5TSY" diet based experiments. The rats died in vain, especially as the study buried the possibility of lethal effects from trans fats.
Peter
This paper, which has been sitting on my hard drive for a few years, surfaced on Facebook recently. I gather it has been cited by someone who’s writing requires more ondansetron to read than I currently possess. I’ll just assume it was some sort of “eat LC and the first smidgin of myocardial hypoxia will finish you off” warning, but I’m just guessing and I have every intention of keeping it that way.
The paper itself is very convincing, well written and the protocol extensively justified. The core findings are that a LC diet impairs insulin signalling, depletes myocardial glycogen and results in massive necrosis during reperfusion after a period of myocardial hypoxia. The basic idea is that the lack of glycogen limits substrate for anaerobic glycolysis and failed insulin signalling both impairs glucose delivery from the perfusate and also fails to deliver a number of highly beneficial insulin effects which are independent of GLUT4 translocation.
This is the fate of the LC myocardium. As one of my co workers might say: Dig the hole, choose the coffin.
Obviously, for a LC eater, this is disturbing. The queue for McDougall-ism is over there.
The first thing which I find slightly disturbing is that, in a trial of the Atkins Diet™ (always mention by name), the rats ate more and were significantly heavier than the control rats, within two weeks. I’m not totally certain if I remember correctly, but I thought that the Atkins Diet was used for weight LOSS, not weight gain. Perhaps the authors might have been a little disturbed by this finding too, but apparently it doesn’t need mention. Hmmm.
The Atkins™ like diet is TestDiet 5TSY, no longer manufactured. Table I is abstracted by the authors from the full formula provided by Purina and supplies information on a need-to-know basis.
So we know total fat, saturated fat and that “The diets have the same concentrations of essential fatty acids”, i.e. we don’t quite know what the diets were made of. But Purina still will email you a pdf (very promptly) and that gives you this:
There is no mention of Crisco™ by name and no information about the trans fat content, but can you guess how much "Vegetable shortening" is in the control diet? Oh, you guessed!
I know it seems stupid to say this, but if you want to lose weight and/or survive a heart attack, for goodness sake do not choose Crisco or its equivalent as 20% of your calorie intake.
Now, this study does not tell us that a low carbohydrate diet is is good or bad for surviving a heart attack, it's simply not possible to pull that information out due to the lack of control of the variables in the diets. One can only wonder whether the formula of the Atkins Rodent Diet was specifically developed to cause metabolic problems or that somewhere along the line the original Atkins diet suggested a generous consumption of trans fat based vegetable shortening. Maybe I missed this.
I guess you could leave it there and say don't eat vegetable shortening, but there are a whole stack of follow on ideas to this study. There is MASSIVE cardiac necrosis in the LC group of rats. If it is the trans fats, how do they cause this?
One very interesting aspect of the study is the pre-ischaemia depletion of glycogen. These rats are not in ketosis. The doubling of ketone levels from 0.3mmol/l to 0.6mmol/l may well be statistically significant, but is not biologically significant. To go back, yet again, to Veech et al, we need around 5.0mmol/l of mixed ketone bodies to completely replace the insulin signalling system. You can't sidestep insulin resistance with 0.6mmol/l B-OHB. You simply cannot get rats in to a functional level of ketosis with protein at 30% of calories and carbohydrate at 12%. You need carbs near zero and protein limited to < 10% of calories to have rats in nutritional ketosis. Some papers limit protein to < 5% of calories, with minimal carbohydrate.
A period of starvation for rats on the control chow would have tested the hypothesis that it was a lack of glycogen which damaged the myocardium under hypoxia.
My own idea is that the glycogen depletion might be a surrogate for insulin resistance rather than carbohydrate restriction. We have long known that trans fatty acids from partially hydrogenated vegetable oils induce insulin resistance (though not in every study!).
So the next thought is: What sort of insulin resistance?
Are we thinking about an excess of superoxide from complex I, to imitate post prandial hyper caloric insulin resistance? Or are we thinking about the insulin resistance of fasting, when uncoupling results in blunted insulin signalling combined with high oxygen consumption and limited ATP generation?
I like the second idea when applied to this study.
If trans fatty acids, which are structurally similar but not quite identical to saturated fatty acids, allow persistent low level uncoupling outside of the physiological role of the normally structured FFAs it is interesting to speculate that they may continue to allow uncoupling when uncoupling should be utterly and totally banned.
The consequences would be decreased ATP production per unit oxygen consumed. A bit like the findings in Table 3 of this paper. The last column gives you the ATP generated per unit O2 consumed. This would be a disaster under hypoxic conditions. The IF line uses industrial trans fats. The soleus muscle is the mitochondrial dependent one cf the tibialis muscle..
ASIDE: It's worth noting that cytoplasmic ATP is a marked inhibitor of uncoupling but that this is easily overcome by adequate levels of mitochondrial ATP binding within the UCP pore from the end which does not produce a conformational change. Figure 7 gives the details but the main text is really clever stuff (without an agenda, as far as I can see). I'll blog about this paper sometime.
Under this control system the occurrence of falling mitochondrial ATP levels should allow cytoplasmic ATP to immediately shut down the normal uncoupling associated with ketogenic eating and maximise coupled ATP production per unit oxygen when this is needed. The interesting question is whether the uncoupling suspected of transfats persists under low mitochondrial ATP levels. END ASIDE.
You could speculate for hours about what trans fats may or may not do.
This is a very fertile area for idea generation. Ultimately, we don't know what a LC diet based on real Food would do to ischaemic damage in the heart. Maybe it will be as bad as a trans fat based LC (weight gain inducing) diet, maybe less severe.
We are never going to find out what Food does by using "AIN-93G Atkins/Rodent 5TSY" diet based experiments. The rats died in vain, especially as the study buried the possibility of lethal effects from trans fats.
Peter
Monday, June 16, 2014
Slim mice which don't fart
Germ free mice are quite interesting. I suppose that the first thing we can say about them is that they don’t have any bacterial fermentation in their gut to produce flatus. As a side issue of some interest is that none of their intestinal mucosal cells ever sees any acetate, butyrate or propionate derived from microbial fermentation of fibre in the gut. They seem rather happy that way. You could also say the same about their liver, it too never sees any SCFA bacterial fermentation products. They stay slim.
Ancient history tells us that germ free mice live rather longer than their more flatulent counterparts. I find this quite interesting.
Of course longevity is a relative term and perhaps ought to be qualified a little. We should actually say that they live longer than conventional mice, provided you feed them. Not feeding germ free mice is quite bad for them, they die under starvation significantly sooner than conventional mice do, at higher bodyweight and with more fat reserves.
That was the state of our knowledge at the end of the last century.
In more recent years the mechanisms for this failure to cope with starvation has become a little clearer. Germ free mice are insulin sensitive. They stay that way pretty much whatever you do to them in terms of diet. They stay that way even if you starve them. That, in terms of survival, is a booboo.
There are at least two techniques available via the gut microbiota which might improve the ability to survive starvation and which are gifted to germ free mice by smearing them with pooh from a conventional mouse. One is endotoxin, a subject I suspect I will come back to. The current one, for this post, is short chain fatty acids.
Bacterial fermentation of fibre produces those miracle agents of gut health and general goodness; acetate, butyrate and propionate. These act through a G protein coupled receptor on enterendocrine cells to promote fat storage. This is the Gpr41 receptor. Needless to say the enteroendocrine cells are the same cells which secrete FIAF, as in the FIAF series of posts. The two are possibly related. Given a little effort we could, by looking at conventionalisation of germ free mice, make a good guess about how much of a mouse’s fat belongs to the mouse and how much belongs to its gut microbiota.
So the fermentation products of bacteria promote fat storage when germ free mice are conventionalised. But no one seems to think that a lack of fat was the reason for germ free mice dying sooner under starvation. So what is the other effect of SCFAs, other than a bigger butt?
The gift of ketosis. Germ free mice are crap at ketosis. It’s not that they can’t do it, it seems to be more like a lack of practice. Acetate and butyrate are particularly ketogenic and hit all sorts of signalling systems in the liver to up regulate ketone generation in conventional mice. Of course germ free mice on standard crapinabag never send acetate to their liver, so their liver never up-regulates the correct PPARs to do ketosis. Putting germ free mice on to a deeply, deeply ketogenic diet teaches them how to make ketones and they become rather good at it.
Under starvation the myocardium of a germ free mouse continues to metabolise glucose, despite free fatty acids being available. Crawford et al consider the myocardium to be fairly representative of many of the glucose using organs in the body. They view ketones as an alternative energy source to glucose under starvation. While no one would argue with this, the possibility which fascinates me is that ketones are turning off glycolysis to spare glucose for the brain when fatty acids alone don’t do this. They appear to be a core component of physiological insulin resistance.
Whether these ketogenic germ free mice are able to extend their time of death to that of starved conventional mice is not a question which any modern ethics committee will allow you to answer today, unless you have a damned good reason. But I suspect the answer is yes.
Peter
Addendum. If you accept that perhaps SCFAs expand your butt via Gpr41 (we are not, after all, germ free mice freshly smeared with conventional mouse faeces) guess which metabolite is a direct antagonist to SCFAs at Gpr41? Clue, it's that the beta hydroxylated derivative of butyric acid. I love stuff that makes sense.
Ancient history tells us that germ free mice live rather longer than their more flatulent counterparts. I find this quite interesting.
Of course longevity is a relative term and perhaps ought to be qualified a little. We should actually say that they live longer than conventional mice, provided you feed them. Not feeding germ free mice is quite bad for them, they die under starvation significantly sooner than conventional mice do, at higher bodyweight and with more fat reserves.
That was the state of our knowledge at the end of the last century.
In more recent years the mechanisms for this failure to cope with starvation has become a little clearer. Germ free mice are insulin sensitive. They stay that way pretty much whatever you do to them in terms of diet. They stay that way even if you starve them. That, in terms of survival, is a booboo.
There are at least two techniques available via the gut microbiota which might improve the ability to survive starvation and which are gifted to germ free mice by smearing them with pooh from a conventional mouse. One is endotoxin, a subject I suspect I will come back to. The current one, for this post, is short chain fatty acids.
Bacterial fermentation of fibre produces those miracle agents of gut health and general goodness; acetate, butyrate and propionate. These act through a G protein coupled receptor on enterendocrine cells to promote fat storage. This is the Gpr41 receptor. Needless to say the enteroendocrine cells are the same cells which secrete FIAF, as in the FIAF series of posts. The two are possibly related. Given a little effort we could, by looking at conventionalisation of germ free mice, make a good guess about how much of a mouse’s fat belongs to the mouse and how much belongs to its gut microbiota.
So the fermentation products of bacteria promote fat storage when germ free mice are conventionalised. But no one seems to think that a lack of fat was the reason for germ free mice dying sooner under starvation. So what is the other effect of SCFAs, other than a bigger butt?
The gift of ketosis. Germ free mice are crap at ketosis. It’s not that they can’t do it, it seems to be more like a lack of practice. Acetate and butyrate are particularly ketogenic and hit all sorts of signalling systems in the liver to up regulate ketone generation in conventional mice. Of course germ free mice on standard crapinabag never send acetate to their liver, so their liver never up-regulates the correct PPARs to do ketosis. Putting germ free mice on to a deeply, deeply ketogenic diet teaches them how to make ketones and they become rather good at it.
Under starvation the myocardium of a germ free mouse continues to metabolise glucose, despite free fatty acids being available. Crawford et al consider the myocardium to be fairly representative of many of the glucose using organs in the body. They view ketones as an alternative energy source to glucose under starvation. While no one would argue with this, the possibility which fascinates me is that ketones are turning off glycolysis to spare glucose for the brain when fatty acids alone don’t do this. They appear to be a core component of physiological insulin resistance.
Whether these ketogenic germ free mice are able to extend their time of death to that of starved conventional mice is not a question which any modern ethics committee will allow you to answer today, unless you have a damned good reason. But I suspect the answer is yes.
Peter
Addendum. If you accept that perhaps SCFAs expand your butt via Gpr41 (we are not, after all, germ free mice freshly smeared with conventional mouse faeces) guess which metabolite is a direct antagonist to SCFAs at Gpr41? Clue, it's that the beta hydroxylated derivative of butyric acid. I love stuff that makes sense.
Sunday, June 15, 2014
Cholesterol: Do chylomicrons clog your arteries? (2)
I'll keep this brief.
The comments on the last post are awash with people trying to help my resident lipophobe out of his lipophobia. This is admirable but misguided and ultimately doomed.
Many years ago I looked at the Copenhagen Heart Study. This observational study generated two hypotheses. One is that chylomicrons kill you. That seems enough for a lipophobe and, if you have this mindset, for goodness sake add carbs to your diet and avoid post prandial hyperlipidaemia. The choice is yours. Go for it. The lack of stress will help you no end.
My own hypothesis is that elevated post prandial triglycerides, here in a population on a mixed diet, is a surrogate for insulin resistance. That is rather similar to an elevated HbA1c under the same circumstances. These are both superficial markers for a failure of complex I of the electron transport chain to effectively deal with the amount of NADH being provided by processing of the diet.
The solution from a Hyperlipid point of view is to concentrate on supplying FADH2 to the ETC via beta oxidation of maximally saturated fatty acids and to minimise NADH input via chronic normoglycaemia, possibly assisted by the inhibition of glycolysis by ketones.
I have no interest in converting lipophobes to lipophiles. We all have our problems, we just have to live with them.
But some gems are coming out of chylomicrons and endotoxin reading. I especially enjoyed this one (among many), all quotes from the same paper:
"Therefore, on the basis of current information, lipoproteins modulate the host response to endotoxin by inhibiting the activation of macrophages, monocytes, and other LPS-responsive cells; promoting the catabolism of LPS by the hepatic parenchymal cells; and inhibiting the response of hepatocytes to pro inflammatory stimuli"
"Early in the course of this work, we found that chylomicron increases the clearance of LPS by the liver while decreasing overall TNF-α production"
"These most recent studies are focused on cells that express the low-density lipoprotein receptor and are critical to the innate immune response to infection, including adrenocortical cells and vascular endothelial cells. The postulated series of events, whereby a foreign molecule (i.e., LPS) serves to both trigger and attenuate a programmed cellular stress response, is unprecedented"
My emphasis.
Now, a functional LDL-C receptor is utterly necessary for the anti-inflammatory effect of the chylomicron/endotoxin complex. If someone's ears don't prick up on this one they have clearly never heard of homozygous familial hypecholesterolaemia. The focus on elevated lipids (sigh) might just have missed the core of the problem, which is the failure to internalise lipoproteins. Interesting idea? It certainly is to me.
Peter
The comments on the last post are awash with people trying to help my resident lipophobe out of his lipophobia. This is admirable but misguided and ultimately doomed.
Many years ago I looked at the Copenhagen Heart Study. This observational study generated two hypotheses. One is that chylomicrons kill you. That seems enough for a lipophobe and, if you have this mindset, for goodness sake add carbs to your diet and avoid post prandial hyperlipidaemia. The choice is yours. Go for it. The lack of stress will help you no end.
My own hypothesis is that elevated post prandial triglycerides, here in a population on a mixed diet, is a surrogate for insulin resistance. That is rather similar to an elevated HbA1c under the same circumstances. These are both superficial markers for a failure of complex I of the electron transport chain to effectively deal with the amount of NADH being provided by processing of the diet.
The solution from a Hyperlipid point of view is to concentrate on supplying FADH2 to the ETC via beta oxidation of maximally saturated fatty acids and to minimise NADH input via chronic normoglycaemia, possibly assisted by the inhibition of glycolysis by ketones.
I have no interest in converting lipophobes to lipophiles. We all have our problems, we just have to live with them.
But some gems are coming out of chylomicrons and endotoxin reading. I especially enjoyed this one (among many), all quotes from the same paper:
"Therefore, on the basis of current information, lipoproteins modulate the host response to endotoxin by inhibiting the activation of macrophages, monocytes, and other LPS-responsive cells; promoting the catabolism of LPS by the hepatic parenchymal cells; and inhibiting the response of hepatocytes to pro inflammatory stimuli"
"Early in the course of this work, we found that chylomicron increases the clearance of LPS by the liver while decreasing overall TNF-α production"
"These most recent studies are focused on cells that express the low-density lipoprotein receptor and are critical to the innate immune response to infection, including adrenocortical cells and vascular endothelial cells. The postulated series of events, whereby a foreign molecule (i.e., LPS) serves to both trigger and attenuate a programmed cellular stress response, is unprecedented"
My emphasis.
Now, a functional LDL-C receptor is utterly necessary for the anti-inflammatory effect of the chylomicron/endotoxin complex. If someone's ears don't prick up on this one they have clearly never heard of homozygous familial hypecholesterolaemia. The focus on elevated lipids (sigh) might just have missed the core of the problem, which is the failure to internalise lipoproteins. Interesting idea? It certainly is to me.
Peter
Thursday, June 12, 2014
Endotoxin Absorption on a High Fat Diet
Housekeeping: Endotoxin is always of some interest to an anaesthetist and the link between endotoxin uptake and high fat diets has been in my thoughts somewhat in recent months. This probably ties in to resistant starch in some way, so the next few posts will probably be random doodles around this subject. And maybe why the Protons thread is fairly well out of data to extend it and another look at brain metabolism, particularly why it doesn't run on fat (or glucose much of the time), all need posts as the time becomes available. Plus Toxic forwarded me a link on B12/folate guidelines which can be downloaded here. As she says, medics may well not read these sorts of guidelines but informed patients certainly should. OK here is the post as was planned.
Endotoxin is nasty stuff. If we want some sort of idea of how nasty it is we just have to look at this juicy quote from Hansen et al from Denmark:
“Harte et al. have reported levels of endotoxin to be between 3.3 and 14.2 EU/mL, but these concentrations are known to increase levels of tumor necrosis factor α (TNFα) in plasma and induce a massive inflammatory response in humans”.
The context of the quote is that these folks are commenting on a paper by Harte et al which described the above levels of endotoxin as being found in human volunteers, which should be nearly lethal. The victims, poor folks, had been coerced in to drinking the sort of amount of whipping cream which might be a lunchtimee snack for myself. And they survived. As do I after such an insult (so far).
What galls Hansen appears to be complete lack of any sort of an inflammatory response to this lethal chylomicron/endotoxin cocktail coursing through the bloodstream of the lipophiles (assuming they enjoyed being paid to drink the cream).
In every group studied, from normal through obese and IGT to diabetic, TNFa falls after a decent glass of whipping cream, albeit in a statistically non significant manner. Endotoxin levels go up, often dramatically and highly significantly.
It’s all in Table 2:

Even in the deepest cesspits of nutritional research no one really expects a half a pint of cream to produce lethal endotoxaemia within a few hours of drinking it. In their response to the comment, which implies that they can’t measure endotoxin correctly, Harte are polite and point out that there is a world of difference between an iv bolus of neat endotoxin (not recommended) and the absorption of endotoxin from the gut in the presence of chylomicrons (welcome to my world).
There are a number of papers showing that apolipoprotein B containing lipid particles are markedly protective against endotoxin, albeit mostly in mouse models. Quite why an oral lipid bolus should automatically load chylomicrons with endotoxin in healthy individuals is another of those fascinating questions which may need a little thought but I think we can reliably say that it would have been eliminated if there is no benefit.
I wrote a post, several years ago, where Greve’s group demonstrated a marked protective effect of a gastric lipid gavage against the effects of haemorrhagic shock, the effects of which are largely related to loss of gut wall integrity, bacterial translocation and endotoxin uptake.
In the high fat world, endotoxin uptake is not what it seems. Lipoproteins neutralise and carry endotoxin. If you need systemic inflammation to maintain research funding you need to be a little more McDevious.
The above little exchange came to mind because Zachary mentioned (in comments on the last post) a study at his university recruiting victims to demonstrate endotoxin uptake after a high fat meal WITH inflammation. You can, of course do this. Technique: "The diet is foods such as TV dinners, meat and cheese". This may be good for getting the right result to secure subsequent funding but it obliterates the fact that a single high fat meal of unadulterated whipping cream is both anti inflammatory and might even improve insulin sensitivity, in the acute setting at least. Just skip the Egg McMuffins.
Peter
Endotoxin is nasty stuff. If we want some sort of idea of how nasty it is we just have to look at this juicy quote from Hansen et al from Denmark:
“Harte et al. have reported levels of endotoxin to be between 3.3 and 14.2 EU/mL, but these concentrations are known to increase levels of tumor necrosis factor α (TNFα) in plasma and induce a massive inflammatory response in humans”.
The context of the quote is that these folks are commenting on a paper by Harte et al which described the above levels of endotoxin as being found in human volunteers, which should be nearly lethal. The victims, poor folks, had been coerced in to drinking the sort of amount of whipping cream which might be a lunchtimee snack for myself. And they survived. As do I after such an insult (so far).
What galls Hansen appears to be complete lack of any sort of an inflammatory response to this lethal chylomicron/endotoxin cocktail coursing through the bloodstream of the lipophiles (assuming they enjoyed being paid to drink the cream).
In every group studied, from normal through obese and IGT to diabetic, TNFa falls after a decent glass of whipping cream, albeit in a statistically non significant manner. Endotoxin levels go up, often dramatically and highly significantly.
It’s all in Table 2:
Even in the deepest cesspits of nutritional research no one really expects a half a pint of cream to produce lethal endotoxaemia within a few hours of drinking it. In their response to the comment, which implies that they can’t measure endotoxin correctly, Harte are polite and point out that there is a world of difference between an iv bolus of neat endotoxin (not recommended) and the absorption of endotoxin from the gut in the presence of chylomicrons (welcome to my world).
There are a number of papers showing that apolipoprotein B containing lipid particles are markedly protective against endotoxin, albeit mostly in mouse models. Quite why an oral lipid bolus should automatically load chylomicrons with endotoxin in healthy individuals is another of those fascinating questions which may need a little thought but I think we can reliably say that it would have been eliminated if there is no benefit.
I wrote a post, several years ago, where Greve’s group demonstrated a marked protective effect of a gastric lipid gavage against the effects of haemorrhagic shock, the effects of which are largely related to loss of gut wall integrity, bacterial translocation and endotoxin uptake.
In the high fat world, endotoxin uptake is not what it seems. Lipoproteins neutralise and carry endotoxin. If you need systemic inflammation to maintain research funding you need to be a little more McDevious.
The above little exchange came to mind because Zachary mentioned (in comments on the last post) a study at his university recruiting victims to demonstrate endotoxin uptake after a high fat meal WITH inflammation. You can, of course do this. Technique: "The diet is foods such as TV dinners, meat and cheese". This may be good for getting the right result to secure subsequent funding but it obliterates the fact that a single high fat meal of unadulterated whipping cream is both anti inflammatory and might even improve insulin sensitivity, in the acute setting at least. Just skip the Egg McMuffins.
Peter
Saturday, May 31, 2014
HbA1c: Low glucose and acid (palmitic)
If anyone would like to combine a look at the pathology of low blood glucose levels with the psychology of diabetes research they might do worse than to look at this paper.
Hypoglycaemia is bad, very bad. At the vascular endothelium level and at the mitochondrial delta psi level. The sort of low levels of blood glucose frequently visited by people like Steve Cooksey and myself are a complete disaster. The group is looking at levels of blood glucose that I can easily achieve by skipping a couple of meals combined with a little mild aerobic exercise, walking or cutting the lawn. I’ve seen 2.7mmol/l on my (admittedly relatively inaccurate) Freestyle Lite. I felt fine at the time and I did nothing to adjust my blood glucose level before having supper a few hours later...
Perhaps I should be carb loading, just a little, to avoid hypoglycaemia induced damage? However this might drop my free fatty acid levels, which I would prefer to avoid.
The abstract of any paper is word limited and the role of FFAs in the findings don't get a mention, so you might assume FFAs have no significant bearing on hypoglycaemic injury.
The discussion section of the paper tends to confirm this:
“Our findings with respect to fatty acid and L-carnitine suggest mechanistically that free fatty acid utilization in vivo would not be sufficient to suppress LG induced NO suppression and excessive mitochondrial superoxide production…”
They seem pretty certain, but do they have data to support this? Flicking to the results section we see that the group looked at a palmitate with L-carnitine combination and found that it worked beautifully:
“We found that combined treatment of endothelial cells with 1% palmitate and L-carnitine reduced LG-induced mitochondrial superoxide production to normal levels (Supplemental Figure IIIa)”
Want the picture?

I think we will all agree that the combination of palmitate with L-carnitine completely, totally normalises superoxide production in the low glucose cells and was utterly harmless in the normal glucose cells. The graph speaks against the discussion.
This normalisation of superoxide generation appears to have been too much to bear, the concept that palmitic acid might be beneficial, so they separated the palmitate from the L-carnitine and tried them separately.
“mitochondrial superoxide production increased under LG conditions with the addition of 1% palmitate while L-carnitine alone returned mitochondrial superoxide levels to those similar to the NG condition (Supplemental Figure IIIb)”
Now, this sticks in my craw. This is wrong. Completely.
Here is Supplemental figure IIIb.

Any reading with an * above it is higher than the normal glucose (NG) reading. This includes the low glucose plus L-carnitine. The low glucose with carnitine value is greater than the normal glucose value, p < 0.05.
There is, however, a crucifix above the LG+LC. The superoxide production here is significantly less than that in the LG group with added neat palmitate, that’s the meaning of the crucifix. But because of the asterix we KNOW that the superoxide here is still higher than the normal glucose value, p < 0.05.
Stating that
“L-carnitine alone returned mitochondrial superoxide levels to those similar to the NG condition”
would require that you (could) remove the asterisk from the low glucose plus carnitine column.
You can't.
You might just as well say the normal glucose plus palmitate (NG+palm) is "similar to the NG condition". Which they don't. Asterisk.
Sigh. End rant.
There are many other things which are both good and bad about this paper. But, as a lipophile living in what sometimes feels like a sea of lipophobia, it really ticks me off to see frankly incorrect and unsupported statements like this. I still scratch my head about motive.
It’s quite clear that palmitate/L-carnitine normalised the excess superoxide production of low glucose completely. I have lots of palmitate in my blood stream, though perhaps not the 1% chemical grade palmitic acid used here! I've probably got reasonable amounts of L-carnitine in my cells too, being a moderate meat eater and highly fat adapted. But the big giggle would really have been to use ketones in the study. I have ketones. As Veech pointed out, many years ago now, ketones can completely replace the whole glucose/insulin metabolic pathway in an isolated rat myocardium. Very effectively.
The flip side is that the paper is looking for information about insulin overdose rather than simple low-normal glycaemia. My BG of 2.7mmol/l was in the presence of utterly basal insulin, markedly elevated free fatty acids and modestly elevated ketones. A diabetic on a sugar based diet having a little (or even quite big) accident with their insulin to starch balance will undoubtedly have suppressed FFAs waaaaay before they hypoed and will probably have a metabolism that can't remember what a ketone body is. The metabolic milieu of parenteral insulin overdose is radically different to that of ketogenic eating. You have to engage thought processes before deciding that any blood glucose below 5mmol/l produces graded superoxide overproduction and failure of nitric oxide signalling. It's understandable from a modern diabetologist point of view, just wrong to bury data which do not support your preconceptions.
If non-injecting (and non sulphonylurea popping) folks want to reduce their FFAs and eliminate ketones in order to avoid low-normal glucose levels by eating a few extra grams of carbs, that is absolutely fine. We can all ignore the biochemistry if we so wish, serious researchers do it all the time.
Personally, I still like to have a few ketones around. And rather a lot of free fatty acids.
Peter
Hypoglycaemia is bad, very bad. At the vascular endothelium level and at the mitochondrial delta psi level. The sort of low levels of blood glucose frequently visited by people like Steve Cooksey and myself are a complete disaster. The group is looking at levels of blood glucose that I can easily achieve by skipping a couple of meals combined with a little mild aerobic exercise, walking or cutting the lawn. I’ve seen 2.7mmol/l on my (admittedly relatively inaccurate) Freestyle Lite. I felt fine at the time and I did nothing to adjust my blood glucose level before having supper a few hours later...
Perhaps I should be carb loading, just a little, to avoid hypoglycaemia induced damage? However this might drop my free fatty acid levels, which I would prefer to avoid.
The abstract of any paper is word limited and the role of FFAs in the findings don't get a mention, so you might assume FFAs have no significant bearing on hypoglycaemic injury.
The discussion section of the paper tends to confirm this:
“Our findings with respect to fatty acid and L-carnitine suggest mechanistically that free fatty acid utilization in vivo would not be sufficient to suppress LG induced NO suppression and excessive mitochondrial superoxide production…”
They seem pretty certain, but do they have data to support this? Flicking to the results section we see that the group looked at a palmitate with L-carnitine combination and found that it worked beautifully:
“We found that combined treatment of endothelial cells with 1% palmitate and L-carnitine reduced LG-induced mitochondrial superoxide production to normal levels (Supplemental Figure IIIa)”
Want the picture?
I think we will all agree that the combination of palmitate with L-carnitine completely, totally normalises superoxide production in the low glucose cells and was utterly harmless in the normal glucose cells. The graph speaks against the discussion.
This normalisation of superoxide generation appears to have been too much to bear, the concept that palmitic acid might be beneficial, so they separated the palmitate from the L-carnitine and tried them separately.
“mitochondrial superoxide production increased under LG conditions with the addition of 1% palmitate while L-carnitine alone returned mitochondrial superoxide levels to those similar to the NG condition (Supplemental Figure IIIb)”
Now, this sticks in my craw. This is wrong. Completely.
Here is Supplemental figure IIIb.
Any reading with an * above it is higher than the normal glucose (NG) reading. This includes the low glucose plus L-carnitine. The low glucose with carnitine value is greater than the normal glucose value, p < 0.05.
There is, however, a crucifix above the LG+LC. The superoxide production here is significantly less than that in the LG group with added neat palmitate, that’s the meaning of the crucifix. But because of the asterix we KNOW that the superoxide here is still higher than the normal glucose value, p < 0.05.
Stating that
“L-carnitine alone returned mitochondrial superoxide levels to those similar to the NG condition”
would require that you (could) remove the asterisk from the low glucose plus carnitine column.
You can't.
You might just as well say the normal glucose plus palmitate (NG+palm) is "similar to the NG condition". Which they don't. Asterisk.
Sigh. End rant.
There are many other things which are both good and bad about this paper. But, as a lipophile living in what sometimes feels like a sea of lipophobia, it really ticks me off to see frankly incorrect and unsupported statements like this. I still scratch my head about motive.
It’s quite clear that palmitate/L-carnitine normalised the excess superoxide production of low glucose completely. I have lots of palmitate in my blood stream, though perhaps not the 1% chemical grade palmitic acid used here! I've probably got reasonable amounts of L-carnitine in my cells too, being a moderate meat eater and highly fat adapted. But the big giggle would really have been to use ketones in the study. I have ketones. As Veech pointed out, many years ago now, ketones can completely replace the whole glucose/insulin metabolic pathway in an isolated rat myocardium. Very effectively.
The flip side is that the paper is looking for information about insulin overdose rather than simple low-normal glycaemia. My BG of 2.7mmol/l was in the presence of utterly basal insulin, markedly elevated free fatty acids and modestly elevated ketones. A diabetic on a sugar based diet having a little (or even quite big) accident with their insulin to starch balance will undoubtedly have suppressed FFAs waaaaay before they hypoed and will probably have a metabolism that can't remember what a ketone body is. The metabolic milieu of parenteral insulin overdose is radically different to that of ketogenic eating. You have to engage thought processes before deciding that any blood glucose below 5mmol/l produces graded superoxide overproduction and failure of nitric oxide signalling. It's understandable from a modern diabetologist point of view, just wrong to bury data which do not support your preconceptions.
If non-injecting (and non sulphonylurea popping) folks want to reduce their FFAs and eliminate ketones in order to avoid low-normal glucose levels by eating a few extra grams of carbs, that is absolutely fine. We can all ignore the biochemistry if we so wish, serious researchers do it all the time.
Personally, I still like to have a few ketones around. And rather a lot of free fatty acids.
Peter
Monday, May 26, 2014
HbA1c: Crack vs smack for a lower reading?
Having lived a great deal of my life in Norfolk, I’ve always rather liked the EPIC Norfolk publications. This was the first paper that I found which demonstrated, at the population level, that HbA1c is an excellent marker for your risk of both CVD and all cause mortality.
HbA1c is one of the most simple biomarkers to manipulate. If there was one single biomarker which you might want to modify, HbA1c is the one.
In Norfolk, people don’t.
Here people generally eat CIAB, more politely known as the UK version of the SAD or a mixed diet.
I doubt very much that anyone eats far enough away from “normal” or “unremarkable” macronutrient ratios that it has any significant effect on the distribution of HbA1c in the population. So, if you take a large group of people and set them free to eat whatever they want, HbA1c is probably a surrogate for mitochondrial health, particularly the mitochondria in adipocytes but more probably those throughout the body.
Nobody doubts that hyperglycaemia is damaging. Hyperinsulinaemia appears to problematic in its own right too. But my impression is that whether you succumb to a disease which is the result of hyperglycaemia directly or due to hyperinsulinaemia, this is just the label. The core problem is the underlying failure of mitochondrial function which signifies to a cell that it’s time to shut up shop. How this shows macroscopically, at the whole organism level, is probably up to a host of factors which I find of limited interest.
Sooooooo. Let’s be very specific: I consider, in the general population, that the linear positive relationship between HbA1c and all cause mortality is a reflection of failing mitochondrial health.
HbA1c is also the easiest of biomarkers to fudge, adjust or fake, call it what you will.
While diabetologists agonise over the inexorable progression of diabetes and wet their knickers at clinically insignificant drops in HbA1c on a calorie restricted mixed diet, those of us who eat LC are well aware that the 5% club is wide open to diabetics and the 4% club is not difficult for those of us who learned LC before mitochondrial damage became widespread. Most, although perhaps not all, people can drop their HbA1c to physiological levels with carbohydrate restriction, possibly with modest supplementation using metformin if they are significantly injured.
Dropping your HbA1c is easy. Very easy.
I have been thinking about this for a long time.
The really difficult question is whether an HbA1c of 4.4% achieved through the strict avoidance of post prandial hyperglycaemia is quite the same as an HbA1c of 4.4% achieved by a random chance combination of accidentally reasonable food choices, a functional pancreas, functional muscles, reasonable parental/intrauterine environment and good luck. Plus anything else you care to think of.
The LC eater is producing an effect on HbA1c which has never been investigated on a population basis, certainly not using modern, insulin resistant people who choose to normalise their blood glucose levels through diet. Whether it is as good for health and/or longevity as achieving it accidentally remains to be seen. No one need ask where my suspicions lie, but there are no hard data. There is absolutely no reason to doubt that being insulin resistant while eating A MIXED DIET indicates that you are in trouble, mitochondrially speaking. Sidestepping this with low carbohydrate eating will undoubtedly reduce your risk of those diseases which are a direct result of the hyperglycaemia and hyperinsulinaemia and which become inevitable on a mixed diet if you are insulin resistant. My own feeling is that this step is vital. I also suspect that some degree of reparation in mitochondrial health might be possible with long term near or frankly ketogenic eating.
OK, enough on the EPIC findings and my thoughts about adjusting HbA1c.
The next big step is to leave Norfolk’s EPIC beauty and wander over to the USA and look at the catastrophic association of a low HbA1c with death. As everyone undoubtedly knows, this link came to me from the exchange between Paul Jaminet and Ron Rosedale over safe starches. I was unaware of the study until Paul brought it up as a foil to the EPIC Norfolk findings.
It too, is a nice observational study.
I have to look at the data here in exactly the same way as I have looked at the data from EPIC. What might it mean? These are observational studies, their use is to generate hypotheses. What is the Hyperlipid view of an HbA1c of 2.8% (that is not a typo) on a mixed USA diet?
The easiest question to answer is whether this might be a surrogate for exceptional mitochondrial health. If you assume that the formula for conversion of HbA1c to average blood glucose levels holds true at the 2.8% level of HbA1c (which it obviously doesn’t), it would represent an average blood glucose of 1.0mmol/l. The lady would be dead. So, before we make idiotic assumptions about average blood glucose levels and mitochondrial health here, we had better think very carefully about what, exactly, a very low HbA1c might really mean. We can say for a start that it is not a simple reflection of glycaemia.
By far the most plausible explanation is not that the red blood cells have been exposed to 1.0mmol/l for the last three months, it’s more likely that these red cells have been exposed to a higher level of glucose, possibly somewhat physiological, for a significantly shorter period of time i.e. the red blood cells are young. Always young. They don’t hang around for long enough to become old and glycated before they are lost. Obviously the study group was well aware of this possibility and corrected for it as best they could.
The lowest HbA1c group had the lowest haemoglobin count, the highest red cell width variability (suggestive of regenerative anaemia) and the highest red blood cell volume (each RBC having relatively little haemoglobin). If you don’t think this group contained some seriously anaemic people you’re probably mistaken. Correcting for red blood cell variables in model 2d of Table 2 gave the bottom limit of the 95% confidence intervals closest to 1.0 of all of the adjusted low HbA1c models.
A quick glance at the ferritin levels, paradoxically, shows that these folks also appear to have iron overload. This may or may not be real as the group also contains 11% people sero positive for hepatitis C virus. As ferritin is an acute phase protein its elevation in a hepatitis C patient may be related to inflammation rather than to iron overload. Correcting for iron based parameters (model 2e) resulted in a worse "higher lowest" limit of confidence intervals than the basic correction for age, ethnicity and sex. You have to wonder if this might be due to an incorrect assumption about ferritin levels.
Of course there were four times as many hep C carriers in the low HbA1c group as the reference group. Model 2f tried to correct for this but I doubt they were able to control for those people using iv heroin (glycaemia neutral) vs those on iv crack vs those on oral stimulants (glycaemia lowering). Anorexia and hypoglycaemia are not uncommon under extended periods of recreational stimulant use. This information seems unlikely to be available to allow adjustment in the NHANES III models.
So I see two studies and each generates a different hypothesis. EPIC suggests that progressive mitochondrial failure on a mixed diet shows as raised HbA1c. The second, from NHANES III, is that sub-physiological HbA1c readings correlate to changes other than physiological glycaemia. Many of these changes appear to bad news, and HbA1c here is the messenger, just as it is in Norfolk.
Using this sort of observational data to convince yourself to increase your starch intake may be a mistake. Avoiding iv drug use and the metabolic consequences there-of might be a better approach.
If anyone can achieve an HbA1c of 2.8% by avoiding starch consumption I'll plaster it all over this post as an edit.
Above all of this, in the realms of sensible living, is the fascinating question of whether an HbA1c of 4.4% achieved by VLC eating has the same (positive) health implications as an HbA1c of 4.4% on a mixed diet.
Peter
BTW the EPIC researchers had a look at low HbA1c in Norfolk. Looks like speed is not the preferred recreational drug here. This fits with my experience dealing with the general population where I live.
HbA1c is one of the most simple biomarkers to manipulate. If there was one single biomarker which you might want to modify, HbA1c is the one.
In Norfolk, people don’t.
Here people generally eat CIAB, more politely known as the UK version of the SAD or a mixed diet.
I doubt very much that anyone eats far enough away from “normal” or “unremarkable” macronutrient ratios that it has any significant effect on the distribution of HbA1c in the population. So, if you take a large group of people and set them free to eat whatever they want, HbA1c is probably a surrogate for mitochondrial health, particularly the mitochondria in adipocytes but more probably those throughout the body.
Nobody doubts that hyperglycaemia is damaging. Hyperinsulinaemia appears to problematic in its own right too. But my impression is that whether you succumb to a disease which is the result of hyperglycaemia directly or due to hyperinsulinaemia, this is just the label. The core problem is the underlying failure of mitochondrial function which signifies to a cell that it’s time to shut up shop. How this shows macroscopically, at the whole organism level, is probably up to a host of factors which I find of limited interest.
Sooooooo. Let’s be very specific: I consider, in the general population, that the linear positive relationship between HbA1c and all cause mortality is a reflection of failing mitochondrial health.
HbA1c is also the easiest of biomarkers to fudge, adjust or fake, call it what you will.
While diabetologists agonise over the inexorable progression of diabetes and wet their knickers at clinically insignificant drops in HbA1c on a calorie restricted mixed diet, those of us who eat LC are well aware that the 5% club is wide open to diabetics and the 4% club is not difficult for those of us who learned LC before mitochondrial damage became widespread. Most, although perhaps not all, people can drop their HbA1c to physiological levels with carbohydrate restriction, possibly with modest supplementation using metformin if they are significantly injured.
Dropping your HbA1c is easy. Very easy.
I have been thinking about this for a long time.
The really difficult question is whether an HbA1c of 4.4% achieved through the strict avoidance of post prandial hyperglycaemia is quite the same as an HbA1c of 4.4% achieved by a random chance combination of accidentally reasonable food choices, a functional pancreas, functional muscles, reasonable parental/intrauterine environment and good luck. Plus anything else you care to think of.
The LC eater is producing an effect on HbA1c which has never been investigated on a population basis, certainly not using modern, insulin resistant people who choose to normalise their blood glucose levels through diet. Whether it is as good for health and/or longevity as achieving it accidentally remains to be seen. No one need ask where my suspicions lie, but there are no hard data. There is absolutely no reason to doubt that being insulin resistant while eating A MIXED DIET indicates that you are in trouble, mitochondrially speaking. Sidestepping this with low carbohydrate eating will undoubtedly reduce your risk of those diseases which are a direct result of the hyperglycaemia and hyperinsulinaemia and which become inevitable on a mixed diet if you are insulin resistant. My own feeling is that this step is vital. I also suspect that some degree of reparation in mitochondrial health might be possible with long term near or frankly ketogenic eating.
OK, enough on the EPIC findings and my thoughts about adjusting HbA1c.
The next big step is to leave Norfolk’s EPIC beauty and wander over to the USA and look at the catastrophic association of a low HbA1c with death. As everyone undoubtedly knows, this link came to me from the exchange between Paul Jaminet and Ron Rosedale over safe starches. I was unaware of the study until Paul brought it up as a foil to the EPIC Norfolk findings.
It too, is a nice observational study.
I have to look at the data here in exactly the same way as I have looked at the data from EPIC. What might it mean? These are observational studies, their use is to generate hypotheses. What is the Hyperlipid view of an HbA1c of 2.8% (that is not a typo) on a mixed USA diet?
The easiest question to answer is whether this might be a surrogate for exceptional mitochondrial health. If you assume that the formula for conversion of HbA1c to average blood glucose levels holds true at the 2.8% level of HbA1c (which it obviously doesn’t), it would represent an average blood glucose of 1.0mmol/l. The lady would be dead. So, before we make idiotic assumptions about average blood glucose levels and mitochondrial health here, we had better think very carefully about what, exactly, a very low HbA1c might really mean. We can say for a start that it is not a simple reflection of glycaemia.
By far the most plausible explanation is not that the red blood cells have been exposed to 1.0mmol/l for the last three months, it’s more likely that these red cells have been exposed to a higher level of glucose, possibly somewhat physiological, for a significantly shorter period of time i.e. the red blood cells are young. Always young. They don’t hang around for long enough to become old and glycated before they are lost. Obviously the study group was well aware of this possibility and corrected for it as best they could.
The lowest HbA1c group had the lowest haemoglobin count, the highest red cell width variability (suggestive of regenerative anaemia) and the highest red blood cell volume (each RBC having relatively little haemoglobin). If you don’t think this group contained some seriously anaemic people you’re probably mistaken. Correcting for red blood cell variables in model 2d of Table 2 gave the bottom limit of the 95% confidence intervals closest to 1.0 of all of the adjusted low HbA1c models.
A quick glance at the ferritin levels, paradoxically, shows that these folks also appear to have iron overload. This may or may not be real as the group also contains 11% people sero positive for hepatitis C virus. As ferritin is an acute phase protein its elevation in a hepatitis C patient may be related to inflammation rather than to iron overload. Correcting for iron based parameters (model 2e) resulted in a worse "higher lowest" limit of confidence intervals than the basic correction for age, ethnicity and sex. You have to wonder if this might be due to an incorrect assumption about ferritin levels.
Of course there were four times as many hep C carriers in the low HbA1c group as the reference group. Model 2f tried to correct for this but I doubt they were able to control for those people using iv heroin (glycaemia neutral) vs those on iv crack vs those on oral stimulants (glycaemia lowering). Anorexia and hypoglycaemia are not uncommon under extended periods of recreational stimulant use. This information seems unlikely to be available to allow adjustment in the NHANES III models.
So I see two studies and each generates a different hypothesis. EPIC suggests that progressive mitochondrial failure on a mixed diet shows as raised HbA1c. The second, from NHANES III, is that sub-physiological HbA1c readings correlate to changes other than physiological glycaemia. Many of these changes appear to bad news, and HbA1c here is the messenger, just as it is in Norfolk.
Using this sort of observational data to convince yourself to increase your starch intake may be a mistake. Avoiding iv drug use and the metabolic consequences there-of might be a better approach.
If anyone can achieve an HbA1c of 2.8% by avoiding starch consumption I'll plaster it all over this post as an edit.
Above all of this, in the realms of sensible living, is the fascinating question of whether an HbA1c of 4.4% achieved by VLC eating has the same (positive) health implications as an HbA1c of 4.4% on a mixed diet.
Peter
BTW the EPIC researchers had a look at low HbA1c in Norfolk. Looks like speed is not the preferred recreational drug here. This fits with my experience dealing with the general population where I live.
Tuesday, May 13, 2014
Eat as much starch as you wish
It has been interesting to watch the discussions about safe starches on those rather limited areas of the internet which I frequent. Now, you have to understand that I have always eaten a certain amount of starch. Usually chips deep fried in beef dripping to go with a fairly standard high fat supper. Sweet potatoes and parsnips are the two usual sources. Certainly two or three times a week. I’m not obese and certainly not post-obese, so I have some freedom in this. I’ve never been zero carb though I am very low carb and I run in mild ketosis pretty much all of the time nowadays.
My routes in to LC, other than the initial window through Atkins, were Kwasniewski, Bernstein, Groves and Lutz. Lutz was such a nice chap, certainly in his writing. He was perfectly willing to work at mere LC levels rather than VLC (72g/d was his advice) and he was very up front that LC would not work for everyone. For inflammatory bowel disease (he was a gastroenterologist) he expected progressive improvement over two years, not two weeks. He discussed that people experiencing failure of LC for weight loss might eventually need to go to simple calorie restricted diets (and I guess he knew they would fail on that too). He had an awareness that exposing an immune system, crippled and ineffective through years of hyperglycaemia, to sudden onset normoglycaemia might render if truly functional for the first time in decades and so exacerbate auto immune attack. Being a medic he would just reach for a 5 day prednisolone course as a simple expedient. For MS patients, many of whom respond very well to LC diets, he took them to around 100g/d initially then down to 72g/d after a few weeks. This is old stuff.
For people to convert from any particular macronutrient ratio to what is a modest, rather than very, low carbohydrate diet is fine by me.
What I have found very disturbing is the long list of potentially catastrophic problems reported to be associated with the type of food choices I make, where there is an edge of ketosis present much of the time. I have said before, I like to have a few ketones available.
It is quite possible that I, and virtually every lab rodent ever placed on a ketogenic diet, might be oddities. Or it might be that 12 years on a very low carbohydrate diet is too soon for the scurvy, thyroid deficiency, auto immune attack or glucose deficiency to get me. Perhaps it will happen tomorrow. But imagine how much money you could save on genetically the modified mice needed for auto immunity research if a simple ketogenic diet gave you a virtually free supply.
There is a saying in the medical community (used by those of us with any sense of self questioning) which goes along the lines of “Clinical experience is no guide to therapeutic efficacy”. One has only to look at a cardiologist with a statin prescription in their hand to understand this. My favourite example is that old chestnut from the siege of Turin during 1536 when Paré realised that pouring boiling oil in to gunshot wounds (the Gold Standard medical treatment of the day, to flush out the toxic carbon particles from the gunpowder residue dontchano) was, to put it mildly, a bit of a booboo.
So I had a listen to the AHS panel discussion on safe starches. The first thing I realised was that no one was making a case for metabolically safe starches, the "safe" referred to a lack of specific plant poisons. Metabolic safety seemed to be a given. I rather liked the lady clinician, she sounded just like Lutz. I loved Chris Kessler too, on C. elegans research. The world is full of people who seem to think humans are unique, free from the metabolic constraints which affect mere nematodes. When my own clients query, in wonder, that cats can get Alzheimers, diabetes, hypertension etc, my reply is that there is nothing special about humans. Failure to learn from C. elegans will lead to some interesting booboos, hopefully not as painful as the boiling oil fiasco.
Ron Rosedale was the only person who came over as making any metabolic sense or having any deep understanding of the processes involved in the signalling subsequent to metabolism.
I have an approach to life. I resist insulin. Running on the edge of ketosis, with a major preponderance of long chain saturated fatty acids as metabolic substrate, I expect to be insulin resistant. I am. It is pure physiology. I like to have uncoupled mitochondria running with a relatively low delta psi, high oxygen consumption and low free radical leakage. On isolated occasions, a few times a week, my parsnip chips will spike my blood glucose and delta psi for an hour or so and generate a few extra superoxide/H2O2 molecules above basal levels. I hope that’s enough for generating a decent number of healthy mitochondria. I don't know if I am correct.
So. I think people should feel free to make their own choices about starch intake. I can't see anything convincing in the concerns that very low carb eating will run you in to a host of medical problems. I'm very uncomfortable with the rhetoric. Quote of the year for 2014 comes from Sid Dishes, relating to the rise of auto immunity caused by VLC eating (I'm possibly only approximately accurate): "Hypothyroidism is so 2011". Sid keeps coming up with these lovely phrases on Facebook. Personally I see no need to add extra starches to what I already eat (between 30 and 60g/d of carbs). This is my rut. I like it.
As we all know only too well, we only get one shot at this. As the Red Hot Chili Peppers said “This life is more than just a read-through”.
Peter
My routes in to LC, other than the initial window through Atkins, were Kwasniewski, Bernstein, Groves and Lutz. Lutz was such a nice chap, certainly in his writing. He was perfectly willing to work at mere LC levels rather than VLC (72g/d was his advice) and he was very up front that LC would not work for everyone. For inflammatory bowel disease (he was a gastroenterologist) he expected progressive improvement over two years, not two weeks. He discussed that people experiencing failure of LC for weight loss might eventually need to go to simple calorie restricted diets (and I guess he knew they would fail on that too). He had an awareness that exposing an immune system, crippled and ineffective through years of hyperglycaemia, to sudden onset normoglycaemia might render if truly functional for the first time in decades and so exacerbate auto immune attack. Being a medic he would just reach for a 5 day prednisolone course as a simple expedient. For MS patients, many of whom respond very well to LC diets, he took them to around 100g/d initially then down to 72g/d after a few weeks. This is old stuff.
For people to convert from any particular macronutrient ratio to what is a modest, rather than very, low carbohydrate diet is fine by me.
What I have found very disturbing is the long list of potentially catastrophic problems reported to be associated with the type of food choices I make, where there is an edge of ketosis present much of the time. I have said before, I like to have a few ketones available.
It is quite possible that I, and virtually every lab rodent ever placed on a ketogenic diet, might be oddities. Or it might be that 12 years on a very low carbohydrate diet is too soon for the scurvy, thyroid deficiency, auto immune attack or glucose deficiency to get me. Perhaps it will happen tomorrow. But imagine how much money you could save on genetically the modified mice needed for auto immunity research if a simple ketogenic diet gave you a virtually free supply.
There is a saying in the medical community (used by those of us with any sense of self questioning) which goes along the lines of “Clinical experience is no guide to therapeutic efficacy”. One has only to look at a cardiologist with a statin prescription in their hand to understand this. My favourite example is that old chestnut from the siege of Turin during 1536 when Paré realised that pouring boiling oil in to gunshot wounds (the Gold Standard medical treatment of the day, to flush out the toxic carbon particles from the gunpowder residue dontchano) was, to put it mildly, a bit of a booboo.
So I had a listen to the AHS panel discussion on safe starches. The first thing I realised was that no one was making a case for metabolically safe starches, the "safe" referred to a lack of specific plant poisons. Metabolic safety seemed to be a given. I rather liked the lady clinician, she sounded just like Lutz. I loved Chris Kessler too, on C. elegans research. The world is full of people who seem to think humans are unique, free from the metabolic constraints which affect mere nematodes. When my own clients query, in wonder, that cats can get Alzheimers, diabetes, hypertension etc, my reply is that there is nothing special about humans. Failure to learn from C. elegans will lead to some interesting booboos, hopefully not as painful as the boiling oil fiasco.
Ron Rosedale was the only person who came over as making any metabolic sense or having any deep understanding of the processes involved in the signalling subsequent to metabolism.
I have an approach to life. I resist insulin. Running on the edge of ketosis, with a major preponderance of long chain saturated fatty acids as metabolic substrate, I expect to be insulin resistant. I am. It is pure physiology. I like to have uncoupled mitochondria running with a relatively low delta psi, high oxygen consumption and low free radical leakage. On isolated occasions, a few times a week, my parsnip chips will spike my blood glucose and delta psi for an hour or so and generate a few extra superoxide/H2O2 molecules above basal levels. I hope that’s enough for generating a decent number of healthy mitochondria. I don't know if I am correct.
So. I think people should feel free to make their own choices about starch intake. I can't see anything convincing in the concerns that very low carb eating will run you in to a host of medical problems. I'm very uncomfortable with the rhetoric. Quote of the year for 2014 comes from Sid Dishes, relating to the rise of auto immunity caused by VLC eating (I'm possibly only approximately accurate): "Hypothyroidism is so 2011". Sid keeps coming up with these lovely phrases on Facebook. Personally I see no need to add extra starches to what I already eat (between 30 and 60g/d of carbs). This is my rut. I like it.
As we all know only too well, we only get one shot at this. As the Red Hot Chili Peppers said “This life is more than just a read-through”.
Peter
Wednesday, April 09, 2014
Dr Doug Wallace on mitochondria
This link came to me from Bill Lagakos via Facebook. It covers a lot of ground and is, unavoidably, a little superficial in places when the subject matter is very deep. He mentions his work with mice made heteroplasmic for two different strains of mitochondria, the NZB and the 129. Both work perfectly well as the sole mitochondrial population giving normal mice. Engineering heteroplasmy produces mice with a stack of problems in high energy demand tissues.
I got to this point, about half way through the presentation, before the penny dropped that the speaker was Doug Wallace, group leader of the people who published this paper
There is a more detailed analysis of this aspect in Nick Lane’s comment in the same edition of Cell about why heteroplasmy might be a Bad Thing, especially in high ATP demanding cells.
I spent much of the presentation thinking that there was no mention of nutritional tools for managing mitochondrial heteroplasmy and very little about mitochondrial mutations and ageing but these came up in the Q and As at the end. The nicest question was about intermittent fasting but Wallace immediately threw in a ketogenic diet as a potential technique for clearing out deleterious heteroplasmic mitochondrial sub populations. He also mentioned the ubiquity of low level heteroplasmy and pre ovulatory selection for optimal mitochondrial population in oocytes...
A lot of ground. Much there which makes sense.
Peter
Thursday, March 06, 2014
Would you like soya oil poured over your methionine spiked casein?
Having a great week off work with my daughter, mostly swimming and gardening, so that blogging is off the radar as I'm fairly much off line at the moment. But...
I guess everyone has seen the takedowns of "meat causes cancer" by Longo et al. so no need for me to chime in. One hundred percent guilty of issuing a press release suggesting causation from an observational study. Duh. Hysterical highlight that meat is cancer for the decade 55-65 years of age and then, come your pension (as a bloke), it becomes the elixir of life. Shrug.
In the same Aussie pile of wallanga pertaining to report science there was a slightly more interesting study published by Le Couteur et al using the standard crippled C57BL/6 mouse strain. They fed umpteen macronutrient ratios to umpteen mice and looked at the longevity. Remember, you cannot feed butter to C57BL/6 mice. Saturated fats break their brain, they have problems with their peroxisomes and with their first phase insulin response, so they in no way resemble any but the most unfortunate of human beings. Feeding them saturated fat is out unless you feed them a ketogenic diet. With that said, enjoy the last comment in this quote from the group:
"Although the mice on a high-protein diet ate less and were slimmer, they also had a reduced lifespan and poor heart and overall health.
Those on a high-carbohydrate, low-protein diet ate more and got fat, but lived longest.
The mice that ate a high-fat, low-protein diet died quickest".
Executive summary: Fat = death.
A brief trip to supplementary data gives what the diets were made of:
"Diets varied in content of P (casein and methionine), C (sucrose, wheatstarch and dextrinized cornstarch) and F (soya bean oil)".
The only fat used was soya bean oil. Can I emphasise again, as many times before:
DO NOT CONSUME BULK CALORIES AS PUFA, ESPECIALLY OMEGA-6 PUFA.
A free "did you notice that?" snippet: Protein was "casein and methionine". Methionine restriction has been shown to prevent metabolic syndrome and possibly to extend lifespan. If you wanted to show protein was bad, might you spike casein by adding extra methionine? To complement the PUFA induced fat-badness??? How do these people sleep at night?
Peter
I guess everyone has seen the takedowns of "meat causes cancer" by Longo et al. so no need for me to chime in. One hundred percent guilty of issuing a press release suggesting causation from an observational study. Duh. Hysterical highlight that meat is cancer for the decade 55-65 years of age and then, come your pension (as a bloke), it becomes the elixir of life. Shrug.
In the same Aussie pile of wallanga pertaining to report science there was a slightly more interesting study published by Le Couteur et al using the standard crippled C57BL/6 mouse strain. They fed umpteen macronutrient ratios to umpteen mice and looked at the longevity. Remember, you cannot feed butter to C57BL/6 mice. Saturated fats break their brain, they have problems with their peroxisomes and with their first phase insulin response, so they in no way resemble any but the most unfortunate of human beings. Feeding them saturated fat is out unless you feed them a ketogenic diet. With that said, enjoy the last comment in this quote from the group:
"Although the mice on a high-protein diet ate less and were slimmer, they also had a reduced lifespan and poor heart and overall health.
Those on a high-carbohydrate, low-protein diet ate more and got fat, but lived longest.
The mice that ate a high-fat, low-protein diet died quickest".
Executive summary: Fat = death.
A brief trip to supplementary data gives what the diets were made of:
"Diets varied in content of P (casein and methionine), C (sucrose, wheatstarch and dextrinized cornstarch) and F (soya bean oil)".
The only fat used was soya bean oil. Can I emphasise again, as many times before:
DO NOT CONSUME BULK CALORIES AS PUFA, ESPECIALLY OMEGA-6 PUFA.
A free "did you notice that?" snippet: Protein was "casein and methionine". Methionine restriction has been shown to prevent metabolic syndrome and possibly to extend lifespan. If you wanted to show protein was bad, might you spike casein by adding extra methionine? To complement the PUFA induced fat-badness??? How do these people sleep at night?
Peter
Sunday, February 16, 2014
Protons (35) TFAM-KO revisited
Those adipose tissue TFAM knockout mice need a revisit. They have an engineered, adipose tissue specific, catastrophic injury to complex I. Complex I failure = insulin-independent DNL, so they should be fat. But they are thin, euglycaemic and have excellent glucose tolerance on a GTT. If complex I failure gives obesity, what is going on here?
These mice were one of the core triggers to me for the concept of TCA halting due to the inability of an injured complex I to oxidise NADH. It's very clear that rotenone, at concentrations which inhibit complex I without killing your model, activates de novo lipogenesis as a technique to deal with excess NADH, excess acetyl-CoA and to provide long chain saturated fatty acids to input as FADH2 to the ETC (at ETFdh) and so to run the rest of the ETC downstream of complex I.
Look at the ability of TFAM adipocytes to uptake 2-deoxyglucose with and without insulin. This is what you get:

TFAM mouse adipocytes appear to be VERY insulin sensitive. In fact they can uptake glucose in the complete absence of any insulin, at the same rate that control (Lox) adipocytes can under supra maximal* insulin. They are insulin hypersensitive without needing any insulin...
*In the supplementary methods these adipocytes are treated with 1.25iu/kg to get this graph, but they are in cell suspension after removal and predigestion, so I don't quite see what concentration of insulin was used. I think is a reasonable assumption that the concentration would be supra maximal...
Yet these adipocytes have mitochondria with a delta psi which is profoundly depressed. The photomicrographs show them as being completely f*cked. So enhanced insulin sensitivity is completely counterintuitive.
What do they do with the glucose they so readily take up? They convert it in to lipid (supplementary data Fig2):
*primary isolated adipocytes, real functional cells, supplied with 5mmol/l glucose.
This lipogenesis looks remarkably like the lipogenesis you see in tissue culture cells treated with rotenone (aside; or in post-obese humans during an OGTT). These cells are energy deprived through the inability to utilise NADH. They side step this by converting acetyl-CoA to lipid and then oxidise this lipid through beta oxidation, which is less dependent on the TCA. The cells appear to be insulin sensitive because they are starving. Even without insulin there is a huge glucose uptake. Once given a few GLUT4s by supplying insulin you get supra-huge glucose uptake. I have to wonder if this is simply a concentration gradient effect, hard to say from the paper. The actual number of GLUT4s (up or down in number cf controls) is completely model dependent, if you read around. But the glucose gets converted to lipid. When you look at ATP turnover you see starvation:

*Note, cell culture 3T3-L1 derivatives at 25mmol/l glucose.
Careful here, we have gone from real mice with TFAM KO adipocytes to TFAM suppressed cell lines. Cultured shTf1 have mild knockdown, shTf2 have severe. These are tissue culture cells. They may or may not behave in quite the same manner as cells from a real live slightly broken mouse...
But the message appears to be that the processes to extract energy from glucose need to be up-regulated to the maximum possible.
Back to real broken mice. Their adipocyte mitochondria are uncoupled (i.e. they fail to correctly increase delta psi when ATP-synthase is blocked by oligomycin) and have an elevated oxygen consumption rate when running on succinate (to bypass complex I)

The uncoupling is very interesting. My feeling is that these cells really are uncoupled, really are insulin resistant due to this and that whatever effect insulin has at high doses is taken advantage of, to the maximum amount possible. Hence the term "apparently" insulin sensitive, when actually insulin resistant...
You could suggest that the underlying insulin resistance is reflected in the glycerol release. These cells, which do insulin-independent glucose uptake, also release glycerol in larger amounts than controls. Oh, and look at the palmitate oxidation too:

The FFAs released from their glycerol backbone do not appear to be discharged in to the systemic circulation (in these mice), they get oxidised because the adipocytes are in ATP starvation and can still use ETFdh to generate some ATP. We have considered FFAs and uncoupling before. The ATP situation may not suggest uncoupling as a particularly good idea but FFAs are FFAs and, if insulin is silenced, then uncoupling seems unavoidable.
BTW the control of uncoupling is so fascinating we'll have to come back to it some other time. Needless to say it integrates ADP, ATP, CoQ redox status, cytoplasm:mitochonrial ATP ratio. And free fatty acid availability, of course.
So let's summarise: Complex I damaged adipocytes are greedy for glucose without (or with) insulin. They are concurrently insulin resistant, which limits their ability to store triglycerides. Their size is immaterial. I was driven to this by a paper which demonstrated that adipocytes isolated from sucrose/lard fed mice are all equally insulin resistant, irrespective of their size (what exactly determines basal lipolysis in an interesting question, I'd guess it is not simply size, though it must be related. It's probably very important). Obviously isolated adipocytes are clear of the influence of leptin, the ventromedial hypothalamus and the sympathetic nervous system, which will certainly not be the case in any intact mouse. But, at the level of very core metabolism, this makes sense in the light of macroscopic observations. When adipocytes STOP getting fatter they do so because complex I has broken to an adequate degree. As they cease to enlarge and cease to respond to insulin they release FFAs, without any concern for higher level metabolic signalling from insulin. Systemic elevated FFAs uncouple the rest of the body's mitochondria (outside the CNS) and on we go through IGT to diabetes.
Peter
These mice were one of the core triggers to me for the concept of TCA halting due to the inability of an injured complex I to oxidise NADH. It's very clear that rotenone, at concentrations which inhibit complex I without killing your model, activates de novo lipogenesis as a technique to deal with excess NADH, excess acetyl-CoA and to provide long chain saturated fatty acids to input as FADH2 to the ETC (at ETFdh) and so to run the rest of the ETC downstream of complex I.
Look at the ability of TFAM adipocytes to uptake 2-deoxyglucose with and without insulin. This is what you get:
TFAM mouse adipocytes appear to be VERY insulin sensitive. In fact they can uptake glucose in the complete absence of any insulin, at the same rate that control (Lox) adipocytes can under supra maximal* insulin. They are insulin hypersensitive without needing any insulin...
*In the supplementary methods these adipocytes are treated with 1.25iu/kg to get this graph, but they are in cell suspension after removal and predigestion, so I don't quite see what concentration of insulin was used. I think is a reasonable assumption that the concentration would be supra maximal...
Yet these adipocytes have mitochondria with a delta psi which is profoundly depressed. The photomicrographs show them as being completely f*cked. So enhanced insulin sensitivity is completely counterintuitive.
What do they do with the glucose they so readily take up? They convert it in to lipid (supplementary data Fig2):
*primary isolated adipocytes, real functional cells, supplied with 5mmol/l glucose.
This lipogenesis looks remarkably like the lipogenesis you see in tissue culture cells treated with rotenone (aside; or in post-obese humans during an OGTT). These cells are energy deprived through the inability to utilise NADH. They side step this by converting acetyl-CoA to lipid and then oxidise this lipid through beta oxidation, which is less dependent on the TCA. The cells appear to be insulin sensitive because they are starving. Even without insulin there is a huge glucose uptake. Once given a few GLUT4s by supplying insulin you get supra-huge glucose uptake. I have to wonder if this is simply a concentration gradient effect, hard to say from the paper. The actual number of GLUT4s (up or down in number cf controls) is completely model dependent, if you read around. But the glucose gets converted to lipid. When you look at ATP turnover you see starvation:
*Note, cell culture 3T3-L1 derivatives at 25mmol/l glucose.
Careful here, we have gone from real mice with TFAM KO adipocytes to TFAM suppressed cell lines. Cultured shTf1 have mild knockdown, shTf2 have severe. These are tissue culture cells. They may or may not behave in quite the same manner as cells from a real live slightly broken mouse...
But the message appears to be that the processes to extract energy from glucose need to be up-regulated to the maximum possible.
Back to real broken mice. Their adipocyte mitochondria are uncoupled (i.e. they fail to correctly increase delta psi when ATP-synthase is blocked by oligomycin) and have an elevated oxygen consumption rate when running on succinate (to bypass complex I)
The uncoupling is very interesting. My feeling is that these cells really are uncoupled, really are insulin resistant due to this and that whatever effect insulin has at high doses is taken advantage of, to the maximum amount possible. Hence the term "apparently" insulin sensitive, when actually insulin resistant...
You could suggest that the underlying insulin resistance is reflected in the glycerol release. These cells, which do insulin-independent glucose uptake, also release glycerol in larger amounts than controls. Oh, and look at the palmitate oxidation too:
The FFAs released from their glycerol backbone do not appear to be discharged in to the systemic circulation (in these mice), they get oxidised because the adipocytes are in ATP starvation and can still use ETFdh to generate some ATP. We have considered FFAs and uncoupling before. The ATP situation may not suggest uncoupling as a particularly good idea but FFAs are FFAs and, if insulin is silenced, then uncoupling seems unavoidable.
BTW the control of uncoupling is so fascinating we'll have to come back to it some other time. Needless to say it integrates ADP, ATP, CoQ redox status, cytoplasm:mitochonrial ATP ratio. And free fatty acid availability, of course.
So let's summarise: Complex I damaged adipocytes are greedy for glucose without (or with) insulin. They are concurrently insulin resistant, which limits their ability to store triglycerides. Their size is immaterial. I was driven to this by a paper which demonstrated that adipocytes isolated from sucrose/lard fed mice are all equally insulin resistant, irrespective of their size (what exactly determines basal lipolysis in an interesting question, I'd guess it is not simply size, though it must be related. It's probably very important). Obviously isolated adipocytes are clear of the influence of leptin, the ventromedial hypothalamus and the sympathetic nervous system, which will certainly not be the case in any intact mouse. But, at the level of very core metabolism, this makes sense in the light of macroscopic observations. When adipocytes STOP getting fatter they do so because complex I has broken to an adequate degree. As they cease to enlarge and cease to respond to insulin they release FFAs, without any concern for higher level metabolic signalling from insulin. Systemic elevated FFAs uncouple the rest of the body's mitochondria (outside the CNS) and on we go through IGT to diabetes.
Peter
Monday, February 03, 2014
Dr Ravnskov on statins for primary prevention of CVD
This is more of a Facebook link than a blog post, but hats off to the BMJ as they have recently published some excellent articles in which neat truth appears to be very controversial (if you pedal b*ll*cks for a living).
Now they have published Dr Ravnskov's response to Ebrahim's pro statin for primary prevention paper. It's a nice reply and it's great to see a medical journal giving a voice to sanity. Of course, hats off to Dr R for being that voice.
You can read the letter here.
Peter
Now they have published Dr Ravnskov's response to Ebrahim's pro statin for primary prevention paper. It's a nice reply and it's great to see a medical journal giving a voice to sanity. Of course, hats off to Dr R for being that voice.
You can read the letter here.
Peter
Friday, January 31, 2014
TV Pantomine or the Oxford study
I don't ever watch television (we have no digital decoding box, the TV is for DVDs) and I can't waste the time to watch this performance on the internet.
There are two truisms I love.
First: How can you tell when a politician is lying? You see their lips move.
Equally good: Any semblance of television documentaries to real life is purely accidental.
For a lesson in how to stack a real study against low carb this Oxford group beats any television entertainment hands down. Also, if you ignore the crap from the researchers, there are lots of interesting (pro LC) data in this study, especially the myocardial energetics and body composition.
What they found, and which made the tittle of the paper, was the gem that LCHF eating "impairs cardiac high-energy phosphate metabolism". Baaaad?
What I really liked was that despite "9% lower cardiac PCr/ATP (P< 0.01)" there was "no change in cardiac function".
Ketones. Ketones allow normal cardiac function at reduced PCr/ATP levels. Ketones (from Veech's work) bypass insulin resistance, be that pathological as in Alzheimers or physiological as in very LCHF eating. Cardiac muscle functions well at reduced ATP levels, provided ketones are available.
Feeling like crap in the early stages of ketosis (Atkins Flu™) is common, it's usually at its absolute worst at about a week in to ketogenic eating. This group clearly knew how to time their cognitive testing! They have more of an agenda than I do.
Both the study and the TV drama have the potential to injure people who need ketogenic eating, I guess the TV show more so than the Oxford study, because it is more likely to stop grass roots level defections from Weight Watchers to genuinely healthy fat-based eating.
Peter
There are two truisms I love.
First: How can you tell when a politician is lying? You see their lips move.
Equally good: Any semblance of television documentaries to real life is purely accidental.
For a lesson in how to stack a real study against low carb this Oxford group beats any television entertainment hands down. Also, if you ignore the crap from the researchers, there are lots of interesting (pro LC) data in this study, especially the myocardial energetics and body composition.
What they found, and which made the tittle of the paper, was the gem that LCHF eating "impairs cardiac high-energy phosphate metabolism". Baaaad?
What I really liked was that despite "9% lower cardiac PCr/ATP (P< 0.01)" there was "no change in cardiac function".
Ketones. Ketones allow normal cardiac function at reduced PCr/ATP levels. Ketones (from Veech's work) bypass insulin resistance, be that pathological as in Alzheimers or physiological as in very LCHF eating. Cardiac muscle functions well at reduced ATP levels, provided ketones are available.
Feeling like crap in the early stages of ketosis (Atkins Flu™) is common, it's usually at its absolute worst at about a week in to ketogenic eating. This group clearly knew how to time their cognitive testing! They have more of an agenda than I do.
Both the study and the TV drama have the potential to injure people who need ketogenic eating, I guess the TV show more so than the Oxford study, because it is more likely to stop grass roots level defections from Weight Watchers to genuinely healthy fat-based eating.
Peter
Saturday, January 25, 2014
An aside on psychiatric links from Sid Dishes
Sid has recently put these two excellent links up on her Facebook timeline. The Protons thread is slowly working its way towards probable causes of complex I failure and an awful lot of the basic mitochondrial information comes from papers on Parkinsons disease or Alzheimers disease.
It has been clear for some time that many, many mitochondrial problems, when localised in specific sets of neurons, are categorised as psychiatric illnesses without needing to have a full blown genetic mitochondrial disease at their core. Acquired mitochondrial dysfunction, most likely at complex I, might well be all you need. Have the right SNPs in genes for proteins of the ETC or assorted ion channels might allow you to be allocated schizophrenia, bipolar disorder or major depression as your pigeonhole.
Trying to treating mitochondrial psychosis by tinkering with the superficial knock on effects at the neurotransmitter level will be of limited effectiveness. Altering bioenergetics using an NAD+ precursor or by inducing ketosis might be far more logical approaches.
Enjoy:
The psychiatric presentation of mitochondrial disorders in adults
Nicotinamide, NAD(P)(H), and Methyl-Group Homeostasis Evolved and Became a Determinant of Ageing Diseases: Hypotheses and Lessons from Pellagra.
The second paper is a rather broad brushed picture of evolution, society and NADH. Gross NAD deficiency with adequate calories (pellagra) will mean that there is almost no time for NAD+ to exist before being reconverted to NADH. The high NADH/NAD+ ratio will particularly favour excess superoxide generation at complex I if there is any reverse electron flow from the CoQ couple. It's a pity this paper does't have mtG3Pdh in its diagram of the ETC. An interesting read even if it's full of concepts which the Hyperlipid perspective might question or might invert the causality there-of.
EDIT: I found this one myself and nearly lost it. The perils of PubMed-ing in your lunch break at work and not emailing the link to yourself!
Neuroanatomical Pattern of Mitochondrial Complex I Pathology Varies between Schizophrenia, Bipolar Disorder and Major Depression
END EDIT
Peter
It has been clear for some time that many, many mitochondrial problems, when localised in specific sets of neurons, are categorised as psychiatric illnesses without needing to have a full blown genetic mitochondrial disease at their core. Acquired mitochondrial dysfunction, most likely at complex I, might well be all you need. Have the right SNPs in genes for proteins of the ETC or assorted ion channels might allow you to be allocated schizophrenia, bipolar disorder or major depression as your pigeonhole.
Trying to treating mitochondrial psychosis by tinkering with the superficial knock on effects at the neurotransmitter level will be of limited effectiveness. Altering bioenergetics using an NAD+ precursor or by inducing ketosis might be far more logical approaches.
Enjoy:
The psychiatric presentation of mitochondrial disorders in adults
Nicotinamide, NAD(P)(H), and Methyl-Group Homeostasis Evolved and Became a Determinant of Ageing Diseases: Hypotheses and Lessons from Pellagra.
The second paper is a rather broad brushed picture of evolution, society and NADH. Gross NAD deficiency with adequate calories (pellagra) will mean that there is almost no time for NAD+ to exist before being reconverted to NADH. The high NADH/NAD+ ratio will particularly favour excess superoxide generation at complex I if there is any reverse electron flow from the CoQ couple. It's a pity this paper does't have mtG3Pdh in its diagram of the ETC. An interesting read even if it's full of concepts which the Hyperlipid perspective might question or might invert the causality there-of.
EDIT: I found this one myself and nearly lost it. The perils of PubMed-ing in your lunch break at work and not emailing the link to yourself!
Neuroanatomical Pattern of Mitochondrial Complex I Pathology Varies between Schizophrenia, Bipolar Disorder and Major Depression
END EDIT
Peter
Thursday, January 23, 2014
Macrobiosis, macrobiopathy?
I hope we all remember Barnad's low fat vegan treatment for diabetes. This figure sums it up:

By 74 weeks folks are not looking very well controlled. Certainly not compared to sustained LCHF. Now, if we crop it nicely we get:

Which makes low fat veganism look pretty good, just so long as you limit your study to 12 weeks.
How a bout a macrobiotic diet?
Very low fat, low protein, LOADS of fibre and complex grain based carbs.
Take some diabetics, measure some numbers, feed for three weeks by skilled macrobiotic cooks while teaching macrobiotic cookery, re test at twelve weeks after self preparing food at home for the last nine of those weeks.
Cured?
We don't get HbA1cs in this study but we have the fasting glucose levels, the 2h post prandial levels, the lipid levels and I quite like the blood pressure levels.
Everything improved when you have a real macrobiotic cook serving you. Do it yourself and by twelve weeks things are already starting to fall to pieces:

Two hour post prandial glucose levels are already rising by 3 months. Even Barnard's study preserved glycaemia for longer than this. You might expect triglycerides to be rising too. They are. If you could give a monkeys about cholesterol levels they too are deteriorating:

Blood pressures pre intervention 127/76, 3 weeks 113/69, 12 weeks, ohoh, 118/75.
Of course you could always claim that these folks weren't doing the macrobiotic diet correctly at home.
Me, I think they are metabolically broken and, even before they stop losing weight, they show their broken metabolism by their climbing post prandial (and fasting) glycaemia. By 74 weeks they will be worse off than Barnards failures.
There is no answer for diabetics other than LCHF.
Sometimes things come up in comments which are so good. As Johnny so kindly obliged the person who linked to the macrobiotic study in their request for comments:
"Yes I would like to comment. Please put your keyboard into a potato sack and throw it into the sewer"
Like.
Peter
BTW, if anyone can reconcile Fig 1 and the red line in Fig 2, congratulations!

By 74 weeks folks are not looking very well controlled. Certainly not compared to sustained LCHF. Now, if we crop it nicely we get:

Which makes low fat veganism look pretty good, just so long as you limit your study to 12 weeks.
How a bout a macrobiotic diet?
Very low fat, low protein, LOADS of fibre and complex grain based carbs.
Take some diabetics, measure some numbers, feed for three weeks by skilled macrobiotic cooks while teaching macrobiotic cookery, re test at twelve weeks after self preparing food at home for the last nine of those weeks.
Cured?
We don't get HbA1cs in this study but we have the fasting glucose levels, the 2h post prandial levels, the lipid levels and I quite like the blood pressure levels.
Everything improved when you have a real macrobiotic cook serving you. Do it yourself and by twelve weeks things are already starting to fall to pieces:

Two hour post prandial glucose levels are already rising by 3 months. Even Barnard's study preserved glycaemia for longer than this. You might expect triglycerides to be rising too. They are. If you could give a monkeys about cholesterol levels they too are deteriorating:

Blood pressures pre intervention 127/76, 3 weeks 113/69, 12 weeks, ohoh, 118/75.
Of course you could always claim that these folks weren't doing the macrobiotic diet correctly at home.
Me, I think they are metabolically broken and, even before they stop losing weight, they show their broken metabolism by their climbing post prandial (and fasting) glycaemia. By 74 weeks they will be worse off than Barnards failures.
There is no answer for diabetics other than LCHF.
Sometimes things come up in comments which are so good. As Johnny so kindly obliged the person who linked to the macrobiotic study in their request for comments:
"Yes I would like to comment. Please put your keyboard into a potato sack and throw it into the sewer"
Like.
Peter
BTW, if anyone can reconcile Fig 1 and the red line in Fig 2, congratulations!
Tuesday, January 21, 2014
I like this
From Phillip (Thanks):
Impaired glucose tolerance in low-carbohydrate diet: maybe only a physiological state.
Like, like, like, like, like, like, like, like, like, LIKE!
Peter
Sunday, January 19, 2014
Acipimox and insulin action
I just wanted to put up a brief mention of this paper on acipimox.
Acipimox is an inhibitor of lipolysis. It's essentially useless as a therapy for anything, partly because it is derived from an incorrect paradigm but mostly because it's impossible to get it to work for any extended period of time. It's good for a week though.
So let's take a few diabetics, do some lab work on them, drop their FFAs using acipimox, then repeat their lab work a week later.
Fasting FFAs drop from 563 micromol/l (not actually very high) to 230 micromol/l (verging on pathologically low) and FBG drops from 8.5mmol/l to 7.0mmol/l. All highly significant, statistically.
Does this mean that they are fixed, i.e. they can go out and eat pizza all day and be normoglycaemic?
No.

A diabetic person who drinks 75g of glucose in water will hit a 2h blood glucose of about 16mmol/l. With markedly reduced FFAs they will be graced with a 2h blood glucose of a mere 14mmol/l, which does not look like dropping. They're f*cked, metabolically. Last time I did this (2008ish??) my 2h BG was 3.4mmol/l.
It looks to me as if acipimox removes the normal physiological uncoupling associated with abnormally elevated FFAs and leaves the insulin resistance of a broken set of mitochondria there for all to see.
There is physiological insulin resistance associated with elevated FFAs. Then there is pathological insulin resistance from mitochondrial dysfunction.
Just wanted to say...
Peter
Acipimox is an inhibitor of lipolysis. It's essentially useless as a therapy for anything, partly because it is derived from an incorrect paradigm but mostly because it's impossible to get it to work for any extended period of time. It's good for a week though.
So let's take a few diabetics, do some lab work on them, drop their FFAs using acipimox, then repeat their lab work a week later.
Fasting FFAs drop from 563 micromol/l (not actually very high) to 230 micromol/l (verging on pathologically low) and FBG drops from 8.5mmol/l to 7.0mmol/l. All highly significant, statistically.
Does this mean that they are fixed, i.e. they can go out and eat pizza all day and be normoglycaemic?
No.

A diabetic person who drinks 75g of glucose in water will hit a 2h blood glucose of about 16mmol/l. With markedly reduced FFAs they will be graced with a 2h blood glucose of a mere 14mmol/l, which does not look like dropping. They're f*cked, metabolically. Last time I did this (2008ish??) my 2h BG was 3.4mmol/l.
It looks to me as if acipimox removes the normal physiological uncoupling associated with abnormally elevated FFAs and leaves the insulin resistance of a broken set of mitochondria there for all to see.
There is physiological insulin resistance associated with elevated FFAs. Then there is pathological insulin resistance from mitochondrial dysfunction.
Just wanted to say...
Peter
Thursday, January 16, 2014
Protons (34) Rotenone
So far I've been thinking about insulin resistance as a normal physiological process for regulating the energy input in to each cell, on an individual metabolic needs basis. There is nothing pathological about this process, regulation is essential. To develop insulin resistance in the immediate aftermath of a single 2000kcal meal or during a 5 day fast is exactly what you need to do. Combining the post prandial state with the fasting state is pathological but could be corrected by simply restoring insulin sensitivity to adipocytes, so allowing them to lower free fatty acids when glucose is elevated.
There are times when I would like there to be differing terms for the insulin resistance of the immediate post prandial period, of starvation and of metabolic broken-ness. It would make for a much clearer picture than saying someone is "insulin resistant".
Now I'd like to think about some pathology.
I’m going to start with this paper on rotenone, with thanks to Mike for the full text. I'm not sure how widely rotenone is used nowadays but, in the lab, it is a freely available inhibitor of complex I. I doubt this toxicosis is particularly prevalent in everyday life, I'm more interested in a basic mechanism which we can extend to other injuries of complex I. Adding rotenone to almost any cell line produces this sort of effect on metabolism, here in C2C12 muscle-like cells (which do it best). From Fig4:

Oxygen consumption is depressed, delta psi is depressed and NADH accumulates at the expense of lowered NAD+ levels. BTW, some authors use the NADH/NAD+ ratio, some the inverse. Ah well, it's still badness. These folks talk about "reductive stress".
The interesting point to note from section D is the rise in acetyl-carnitine, a molecule used to export acetyl-CoA from mitochondria to cytoplasm.
If we block the ability of the mitochondria to feed NADH in to the ETC there is no point in turning the TCA because this generates four NADHs for that single FADH2 from succinate dehydrogenase (which still works if you provide succinate exogenously). Instead the acetyl-CoA is exported as acetyl-carninitine or as citrate after combination with oxaloacetate. Once in to the cytoplasm acetyl-CoA is available for de novo lipogenesis. Does it get used for this?
Here are some C2C12 muscle-like cells which are running on a "low" concentration (probably around 5.0mmol/l) of glucose. The ones on the right are also exposed to 5.0 nanomol of rotenone in the complete absence of insulin or any equivalent. Cytoplasm is pale purple, nucleus is dark purple:

The beautiful orange-red staining droplets are lipid, more clearly visible in this enlargement:

How much lipid is deposited? That depends on how much rotenone is used and how long the complex I has been blocked for. From Fig 2:

You can do exactly the same thing by blocking complex I with piericidin A or by knocking down expression of the gene NDUFV1 (codes for a huge chunk of complex I) in C2C12 cells. From Fig 3:

You get the same effect in people with the misfortune to be born with severe defects in complex I; in the affected tissues there is lipid accumulation. By the time you get down to <5% complex one activity there are quite nasty knock on effects on fatty acid oxidation and cell viability, but that's another story.
Very few people would choose to take rotenone nowadays but there are a number of other complex I inhibitors, some of which are quite widely available. Flunarizine, isoniazid and atrazine spring to mind with bisphenyl A as a more general mitochondrial toxin which includes complex I blockade. None are marketed as weight loss supplements.
I see this as a generic mechanism. Complex I dysfunction leads to intracellular lipid accumulation.
What else is interesting?
These cells are being fed with low levels of glucose without insulin. Glucose uptake will be basal and much of the energy from glycolysis will be diverted to lipogenesis. Again, if ox phos is reduced mitochondrial ATP production will be depressed and glycolysis will be the main source of ATP using substrate level phosphorylation. We know lipogenesis is occurring, so we must be deriving NADPH from glycolysis. If we were to add insulin to the culture medium of a rotenone poisoned cell and perhaps increase the glucose level to 25mmol/l, what would happen?
Assuming we can generate enough of a delta psi to allow insulin signalling we would pour glucose in to the cell and down through glycolysis to pyruvate. This gives us a ton of acetyl-CoA in the mitochondria. Complex I is still blocked. What's a cell to do except de novo lipogenesis in its cytoplasm?
As we allow glucose in to a cell, we push de novo lipogenesis. This is a parody of insulin sensitivity. The faster we allow glucose in to a cell, the faster it is lost to lipid. My line of thought is about how well this ability to sequester glucose as lipid might give the impression of being extremely sensitive to insulin, when the actual mitochondria are dysfunctional and should be signalling insulin resistance.
I can't get away from the unarguable fact that post-obese women perform enough DNL to get their RQ > 1.0 on exposure to 75g of glucose in an OGTT.
Perhaps we need to look at the mitochondrial function of people who's "preferred" metabolic state is best achieved by obesity. And how we might damage complex I without mainlining rotenone.
Better hit "post" as things keep getting in the way of extending this particular entry.
Peter
There are times when I would like there to be differing terms for the insulin resistance of the immediate post prandial period, of starvation and of metabolic broken-ness. It would make for a much clearer picture than saying someone is "insulin resistant".
Now I'd like to think about some pathology.
I’m going to start with this paper on rotenone, with thanks to Mike for the full text. I'm not sure how widely rotenone is used nowadays but, in the lab, it is a freely available inhibitor of complex I. I doubt this toxicosis is particularly prevalent in everyday life, I'm more interested in a basic mechanism which we can extend to other injuries of complex I. Adding rotenone to almost any cell line produces this sort of effect on metabolism, here in C2C12 muscle-like cells (which do it best). From Fig4:

Oxygen consumption is depressed, delta psi is depressed and NADH accumulates at the expense of lowered NAD+ levels. BTW, some authors use the NADH/NAD+ ratio, some the inverse. Ah well, it's still badness. These folks talk about "reductive stress".
The interesting point to note from section D is the rise in acetyl-carnitine, a molecule used to export acetyl-CoA from mitochondria to cytoplasm.
If we block the ability of the mitochondria to feed NADH in to the ETC there is no point in turning the TCA because this generates four NADHs for that single FADH2 from succinate dehydrogenase (which still works if you provide succinate exogenously). Instead the acetyl-CoA is exported as acetyl-carninitine or as citrate after combination with oxaloacetate. Once in to the cytoplasm acetyl-CoA is available for de novo lipogenesis. Does it get used for this?
Here are some C2C12 muscle-like cells which are running on a "low" concentration (probably around 5.0mmol/l) of glucose. The ones on the right are also exposed to 5.0 nanomol of rotenone in the complete absence of insulin or any equivalent. Cytoplasm is pale purple, nucleus is dark purple:

The beautiful orange-red staining droplets are lipid, more clearly visible in this enlargement:

How much lipid is deposited? That depends on how much rotenone is used and how long the complex I has been blocked for. From Fig 2:

You can do exactly the same thing by blocking complex I with piericidin A or by knocking down expression of the gene NDUFV1 (codes for a huge chunk of complex I) in C2C12 cells. From Fig 3:

You get the same effect in people with the misfortune to be born with severe defects in complex I; in the affected tissues there is lipid accumulation. By the time you get down to <5% complex one activity there are quite nasty knock on effects on fatty acid oxidation and cell viability, but that's another story.
Very few people would choose to take rotenone nowadays but there are a number of other complex I inhibitors, some of which are quite widely available. Flunarizine, isoniazid and atrazine spring to mind with bisphenyl A as a more general mitochondrial toxin which includes complex I blockade. None are marketed as weight loss supplements.
I see this as a generic mechanism. Complex I dysfunction leads to intracellular lipid accumulation.
What else is interesting?
These cells are being fed with low levels of glucose without insulin. Glucose uptake will be basal and much of the energy from glycolysis will be diverted to lipogenesis. Again, if ox phos is reduced mitochondrial ATP production will be depressed and glycolysis will be the main source of ATP using substrate level phosphorylation. We know lipogenesis is occurring, so we must be deriving NADPH from glycolysis. If we were to add insulin to the culture medium of a rotenone poisoned cell and perhaps increase the glucose level to 25mmol/l, what would happen?
Assuming we can generate enough of a delta psi to allow insulin signalling we would pour glucose in to the cell and down through glycolysis to pyruvate. This gives us a ton of acetyl-CoA in the mitochondria. Complex I is still blocked. What's a cell to do except de novo lipogenesis in its cytoplasm?
As we allow glucose in to a cell, we push de novo lipogenesis. This is a parody of insulin sensitivity. The faster we allow glucose in to a cell, the faster it is lost to lipid. My line of thought is about how well this ability to sequester glucose as lipid might give the impression of being extremely sensitive to insulin, when the actual mitochondria are dysfunctional and should be signalling insulin resistance.
I can't get away from the unarguable fact that post-obese women perform enough DNL to get their RQ > 1.0 on exposure to 75g of glucose in an OGTT.
Perhaps we need to look at the mitochondrial function of people who's "preferred" metabolic state is best achieved by obesity. And how we might damage complex I without mainlining rotenone.
Better hit "post" as things keep getting in the way of extending this particular entry.
Peter
Subscribe to:
Posts (Atom)
