Differential Metabolic Effects of Saturated Versus Polyunsaturated Fats in Ketogenic Diets
I think I worked through this several years ago but didn't have the tools to comment on it at the time. The recall I have is of reading what the high PUFA group ate. Fake bacon, soy nuts, vegetable oil. Lovely. BUT they ended up much more insulin sensitive than the saturated fat group did over 5 days. Higher ketones, lower glucose, lower trigs, higher insulin sensitivity. I had no explanation for this.
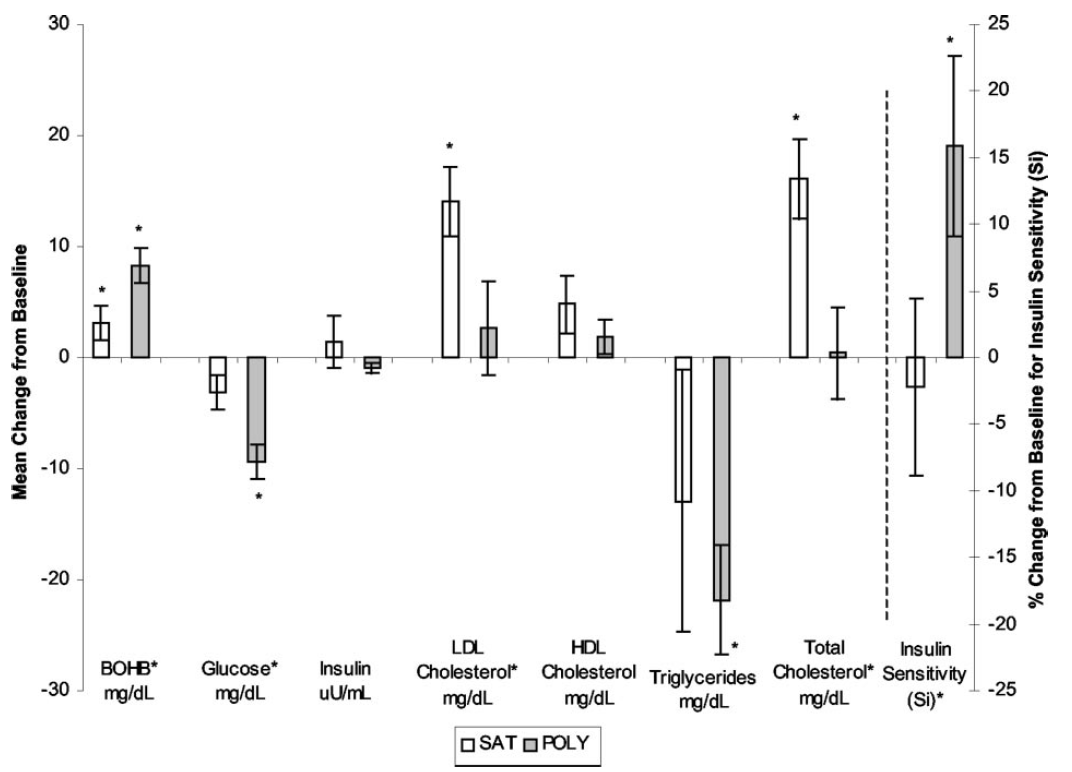
Once you appreciate the Protons concept this is exactly what you would expect. There is continued insulin signalling when there should be physiological insulin resistance. While ever adipocyte size is kept low, via the ketogenic nature of the diet, enhanced insulin sensitivity should persist.
The down side is that glucose metabolism will continue. If your approach to life is to stop using glucose as an energy substrate there is absolutely no need to maintain glycolysis in the face of a ketogenic diet. Simply refusing to listen to insulin is my preferred option. Someone running their metabolism of fats should have minimal need for insulin sensitivity and buying insulin sensitivity at the cost of metabolising linoleic acid, with its daughters 4-HNE and 13-HODE, is not something which appeals to me. But be aware of the study and the joy it will give to the saturophobes...
Peter

27 comments:
I wonder if upping the PUFAs right before a carb refeed would benefit an athlete/carb cycler, or does O-6 just suck worse than the higher sugar in the blood whlle insulin resistant?
I don't know, might help. And of course long term health is slightly down the priority list compared to winning! Hence doping rules!
Peter
I wonder what Gerhard Spiteller and Fred Kummerow would have said, may they rest in peace.
I am in for the Long Term, who needs a medal around the neck if it becomes a noose? Shannon
Peter,meant to ask this before, but what is the protons view of the upset stomach when I drink a pint of double cream, esp. when it's hot outside? :) If I'm a paleo hunter that would be pretty inconvenient :)
I believe you referred to it too.
I doubt a paleo hunter would have access to cream once weaned! But I agree and I think the effect is probably hormonal, possibly cholecystokinin but Im guessing there. I too get the queasiness effect from too much cream in one drink, but I've also done the same with gross overdose of very fatty lamb chops. With access to that amount of fatty meat I wouldn't be hunting!!!!!
Peter
OK, maybe I'm just dense (not 'maybe), but how do we reconcile this:
"Once you appreciate the Protons concept this is exactly what you would expect. There is continued insulin signalling when there should be physiological insulin resistance. While ever adipocyte size is kept low, via the ketogenic nature of the diet, enhanced insulin sensitivity should persist."
Where n-6 keeps insulin low, with this, where n-6 feeding appear to increase IR and insulin goes higher?
"What about health? Here are the numbers which matter, obviously insulin is the one we want to look at..."
http://high-fat-nutrition.blogspot.com/2017/03/trans-fats-vs-linoleic-acid.html
There are situations where reducing n-6 lowers insulin... In vivo in humans.
Is it HNE overwhelming the proton effect of LA?
Hi Tucker, thanks for the comment! Consideration from anyone not as immersed in these ideas as me is much appreciated.
I wouldn’t expect any PUFA to cause acute insulin resistance due to their oxidation, provided adipocyte is size is not allowed to increase. Perhaps unless the levels are very high (triglyceride/heparin infusions and the like) at which time any FFAs can interfere with the ETC directly, the one study which floated this idea had them interacting with the binding site for CoQH2, I think, at the complex III docking site. This was independent of oxidation of the fat. I’ve never been able to find follow on to this, it’s of great interest.
Yes, insulin is very important. If you can keep adipocyte size low then you could potentially reap benefits long term from PUFA. However, as you know, there are core problems with PUFA incorporation on the ETC function if there is any superoxide generation near the cytochrome C anchorage lipids. I wouldn’t expect you can ever stop superoxide generation completely. Once you do generate superoxide, which would be a simple signalling molecule in a saturated fat environment, you can generate 4-HNE (plus others I've not really thought about). I think I’ve seen papers where lipid peroxides such as 4-HNE generate insulin resistance per se.
I almost included the trans fats along with fructose and alcohol as a mediator of inappropriate lipolysis in the current series. Like alcohol, the cost of adipose tissue lipolysis when it’s not appropriate (post prandial) is FFA release with uptake of excess by the liver, to be released later as triglycerides, en rout to visceral adipocytes. I think I was wrong in that post that there might be benefit to trans fats. They might be about as good as alcohol for fat loss, true, but as they are always consumed with carbohydrate we're back in to elevated glucose, insulin and FFAs. Not good all at once.
I feel adipocyte distention is going to be the overwhelming effect of LA…. But I also feel the need to think about the CVD exacerbation in the Sydney Diet Heart Trial and the ilk. As always, there is more than one thing going on.
More discussion very welcome.
Peter
So I think your 3rd paragraph is the key to reconciliation (I broke your paragraph up and added numbers to organize my response):
[4] "Yes, insulin is very important."
[2] "If you can keep adipocyte size low then you could potentially reap benefits long term from PUFA."
[1] "However, as you know, there are core problems with PUFA incorporation on the ETC function if there is any superoxide generation near the cytochrome C anchorage lipids. I wouldn’t expect you can ever stop superoxide generation completely. Once you do generate superoxide, which would be a simple signalling molecule in a saturated fat environment, you can generate 4-HNE (plus others I've not really thought about)."
[3] "I think I’ve seen papers where lipid peroxides such as 4-HNE generate insulin resistance per se."
[1] is the key. I think your Proton model represents the 'normal' behavior, with a normal level of n-6 PUFA in the diet; a non-pathological (pre- might be the appropriate prefix) model. There's an entirely separate pathway, however, which I think drives the pathology when n-6 goes above some threshold.
Stopping superoxide generation is nether possible nor desirable: the system is designed to expect it, and it's a beneficial process in the normal range, which is why exogenous antioxidants are detrimental to adaptation.
Superoxide, while present, is not required for PUFA oxidation. Linoleic Acid and glucose in physiological amounts in water in vitro will auto-oxidise, and generate HNE. Adjacent LA chains in cardiolipin are required for cardiolipin oxidation to occur, tetra-linoleoyl cardiolipin (TLCL) is therefore uniquely susceptible to oxidation. Replace LA with Oleate, and cardiolipin won't oxidize. Iron catalyzes LA oxidation, Cytochrome-C contains an iron atom, and upon exposure to cardiolipin, deforms to expose the iron, thus catalyzing the cardiolipin oxidation. In vitro, if incubated together, Cyt-C will oxidize TLCL until none remains.
Both seem important in vivo. Additionally, they not only generate HNE, they generate singlet oxygen. Although since HNE is 1000x as pro-oxidant as singlet oxygen, it may not matter much. The self-sustaining reaction is seen in ARDS in vivo in humans.
Of course this process destroys the ETC, as it's composed of cardiolipin, and thus the mitochondria, and then the cell. Which explains three universal aspects of chronic disease: mitochondrial dysfunction, apoptosis, and necrosis.
The above is what is called lipid peroxidation or oxidative stress (OsStr).
Superoxide and ROS can't exit the mitochondria, but HNE can.
[2] HNE then induces adipocyte dysfunction and enlargement, and [3] insulin resistance directly, and indirectly through oxidation of LDL.
The latter effect is a fundamental part of our immune system, and induces systemic insulin resistance.
HNE is a systemic toxin, and may also oxidize n-3, thus accounting for HHE levels and HHE induction of IR in vivo.
My wager is that hyperglycemia is a result of OxStr-induced dysregulation of gluconeogenesis, perhaps though mitochondrial dysfunction, perhaps through HNE damage. Hyperglycemia accelerates OxStr, through what I imagine is a mitochondrial ETC mediated upregulation in ROS production, but I haven't been able to figure out the mechanism, but it's a drastic effect.
You have a far better understanding of the ETC than I, so any input would be appreciated in figuring out that bit...
Thoughts?
I have this myopic view which tries to fit macroscopic events in to probable changes in the redox state of the first section of the ETC. I look at what RET should do, what might increase it, what might decrease it, just on an input basis. Does this fit in with the whole animal effect? I think of first level up local damage as an amplifier of the basic ETC effect and I'd put both H2O2 and 4-HNE in as first level up messengers. What they do should do either to amplify a small but necessary local change or be part of negative feedback. I look at more traditional signalling molecules, cytokines etc, as the next level up and hormonal changes as the layer up from there. So I try to see the end result in terms of what happens deep down. The details of the intermediate layers is definitely not something I'm any good at. When I see interleukin 1, 6, 19 or 555 I tend to glaze over and think about what that interlukin represents, what the underlying process might be, deeper down. Correct that and you can throw you cytokine modifiers down the drain. Unless you have ARDS of course. Might be interested then, but that's because the cytokines are going to kill you per se and the system is very broken. Avoiding major trauma is a good idea...
Peter
Tucker:
"Hyperglycemia accelerates OxStr, through what I imagine is a mitochondrial ETC mediated upregulation in ROS production, but I haven't been able to figure out the mechanism, but it's a drastic effect."
Probably my misunderunstanding, but why don't you consider high complex I activity due to high glucose levels?
I'm quite sure you did, but rejected it for some reason?
Yes, high glucose will cause high complex I activity, but why or how does that increase OxStr?
I haven't ever seen a demonstration, what I find says ROS tends to be higher on SFA.
It maybe topographic, as cardiolipin is closer to source...
But I would like tok ow.
Tucker, yes, ROS would be higher on saturates. But the problem is the hyperglycaemia, if glycolysis is raising NADH so driving the mitochondrial glycerophosphate shuttle (to reduce NADH levels and so protect the cell from reductive stress) we will have a marked rise in CoQH2 levels, ripe for RET. If we add saturate derived FADH2 also generating CoQH2 we are really set for RET. Lowering the saturated fat FADH2 will generate less RET as PUFA don't provide nearly as much FADH2. So I view PUFA as protecting against the abnormal state of hyperglycaemia. If the diet was based on sat fat in the first place I don't think the obesity would develop, we wouldn't have insulin resistance from leaking FFAs from adipocytes in defiance of insulin and we would never have the hyperglycaemia.
So I think PUFA protect against RET excess triggered by hyperglycaemia at the cost of obesity and mangling the ETC long term. Which is no solution.
Re topography: Absolutely, if it is beneficial to eliminate a mitochondrion which is generating too much ROS, especially from complex I, I'd be willing to bet that supercomplex assembly puts complex I within ROS distance of cardiolipin. Not seen anything to support this but evolution is not stupid (without punishment)...
Peter
OK. I more-or-less understand that. What I don't understand is how well reconcile that with the paper below. The protocol used promptly destroys the mitochondria (with pictures!). If complex I produced ROS under hyperglycemia, this would make sense...
This paper is a perfect model of late-stage MetSyn, BTW, or human heart failure. Of course that's redundant...
"Brief episode of STZ-induced hyperglycemia produces cardiac abnormalities in rats fed a diet rich in n-6 PUFA"
link: https://t.co/XhH6BomJCH
Tweet: https://t.co/8Mcp130ZQB
Yes, I picked that study up about 9m ago. My wife is involved as the histopathologist assisting two PhDs looking at myocardial failure in cats. Obviously, most modern cats are PUFA/starch fed and clearly develop metabolic syndrome. The PhDs are looking for changes designating hypertrophic cardiomyopathy in post mortem cases and a notable problem is that very few of the cats in the "normal" group are normal. Feeding a cat high PUFA plus starch diet may not be as spectacular as those rats but I think they (the cats) will be chronically intermittently hyperglycaemic on a PUFA background. The rat study has certain problems but is essentially looking at very high glycolysis rates using hyperglycaemia assisted glucose entrance to cells to replace insulin facilitated entrance (i.e. diabetes). As such it is not regulated, neither is the FFA supply, even if the plasma FFAs are not raised (these are fed rather than fasted samples). I would contrast this the a more normal physiological state (non diabetic) where PUFA are supplying too little FADH2 to the ETC to limit insulin signalling in the fed state. The continued insulin signaling will allow continued ingress of calories until eventually the inputs from the low level of FADH2 from PUFA is matched by increased FADH2 from the glycerophosphate shuttle and caloric ingress does stop, admittedly with distended adipocytes. An overwhelming input from unregulated glucose and unregulated PUFA is going to cause pathological RET with no way of stopping it, excess caloric ingress can only be stopped by RET if it is being allowed by insulin in the first place. Limiting the changes that loss of insulin allows cannot be stopped by limiting insulin's action using RET. In this study you just have excess of everything.
Once you get in to this situation all that shows are the problems as PUFA facilitate oxidative damage to lipids in the mitochondria, so badness happens. Diabetes without PUFA will do less damage to the cardiolipins but the possibility of getting obese without PUFA is much smaller, so type 2 diabetes is less likely at all. I think humans can become diabetic with sucrose and flour but it’s nothing like as easy as with PUFA…
Peter
Another afterthought: If they had calorie restricted the PUFA rats (severely) to keep them slim and insulin sensitive, what would have happened after the streptozotocin? Without insulin being present insulin resistance not going to act as a regulator. Insulin resistance will not be able to limit caloric ingress because ingress is not being facilitated by insulin. The mitochondria would still be full of PUFA cardiolipins and shit would still happen, without the obesity or metabolic syndrome.... Might not be as bad because the cardiomyocytes might not be full of lipid droplets waiting to explode in to 4-HNE. The ETC should still fall to pieces.
That's my guess
Peter
Peter,
First of all thank you for your continued blogging.
What about a case of keto diet with decent amount of PUFA and Saturated Fat as well.
Lets say 50/50 ratio.
What would a Protons™ view of that be?
If PUFA fail to generate insulin resistance via RET through Complex I would adding SF correct for it?
Does PUFA simply fail to generate insulin resistance or does it also actively increase Insulin sensitivity?
Thx,
Slobodan
Cunnane's work on the fate of PUFAs helps to explain why they increase insulin sensitivity.
1) there is a high rate of carbon recycling of LA to cholesterol, cholesteryl esters, SFAs, as well as conversion to eicosanoids. So at least 25% of their energy is just unavailable to fuel muscle.
Therefore, the muscle has to keep running on glucose, more than if the fat were say palmitate which does zero carbon recycling.
2) despite this loss to carbon recycling, and no DNL production of LA, there is a relatively high proportion of LA in adipose.
3) if, for whatever reason, LA lowers LDL, there isn't the ApoB to supply TGs to muscle.
LA doesn't generate much ROS in muscle because IT FINDS IT HARD TO GET THERE is my working hypothesis.
Hi Slobodan, I suspect you would end up like F3666 fed mice, slim, basically health but with focal steatohepatitis. If you did manage to elevate your BG I suspect that having an ETC full of polyunsaturated cardiolipins might very unforgiving....
George, an interesting concept. I'm assuming you keep testing it in your head....
Peter
Indeed I do Peter - and came across this in my memory
- when CHO is restricted, and therefore fatty acids must be used by muscles, serum SFAs drop while MUFA stays the same and PUFAs increase.
Sure, DNL drops, but MUFA is also a DNL product. This does make it seem as if SFAs, and the shorter chain ones at that (C14 takes the biggest drop) are being preferentially used.
It's as if C18:1 is just a buffered (less physiologically active) storage form of SFA.
That, George, is a very, very nice observation. I'm just a little bit jealous! Nice...
Peter
Thanks! And here's, finally, the sort of data I was looking for, though it's early days yet in my search.
https://www.sciencedirect.com/science/article/pii/S0005272805002574#fig1
My interpretation is that a fat which, when supplied, is more likely to be oxidised to generate ATP by muscle will suppress the oxidation of glucose more than a fat which is less useful. (there is also the supply issue for LA and caprylic acid in vivo* but in vitro experiments sidestep that). Here, these are the LDL-raising SFAs C12 and C14 for one muscle type, and C12, C14, and C16 for the other, as well as C8. Except for C8 the glucose oxidation and LDL-raising effects match pretty well (but C8 is ketogenic via liver in vivo so needs less transport on lipoproteins).
Insulin -ve states are what we are most interested in. Later they get into uncoupling and O2 consumption - the O2 is pretty level across FFAs, working against the hypothesis, but the uncoupling can explain this; not much use for actually fueling muscle, the relative glucose demand seems to be a clearer guide to this, if I'm not deluding myself.
And of course FADH/NADH and RET provide a mechanism to signal this demand or its absence to the cell surface.
* uptake of MCFA across the leg is minimal (6–9%), from
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC151643/
I also like this - supply of C18:1 increases oxidation of C16 in muscle.
Increased Mitochondrial Fatty Acid Oxidation Is Sufficient to Protect Skeletal Muscle Cells from Palmitate-induced Apoptosis
(Title belongs in "so bleeding obvious it needed saying" basket).
http://www.jbc.org/content/285/47/36818.full.html
Oleate preincubation exerted its protective effect by two mechanisms: (i) in contrast to CPT1mt expression, oleate preincubation increased the channeling of palmitate toward triglycerides, as a result of enhanced diacylglycerol acyltransferase 2 expression, and (ii) oleate preincubation promoted palmitate oxidation through increasing CPT1 expression and modulating the activities of acetyl-CoA carboxylase and AMP-activated protein kinase.
Wow. There are an essentially infinite supply of studies which show palmitate in tissue culture, will explode cells (always glucose content of medium not specified, assume 25mmol/l) but there is a very small subgroup of these garbage studies where even very small amounts of oleate are essentially fully protective against palmitate induced apotosis. You are now picking at the mechanism. Without digging in to the depths of my hard drive my memory suggest that the apoptosis induction is through ceramide formation, not palmitate oxidation. Once I read ceramide I think "higher level signalling" and tend to glaze over. But avoiding ceramide formation by increasing palmitate oxidation, now there you have a LC adaptation....... So cool!
Peter
So two things come to mind -
1) you need at least one MUFA to make a TG in mammalian metabolism. With only C16 in culture, diversion into neutral storage form isn't possible. This one way in which C18:1 can be "buffered storage form of SFA".
2) incubation with 18:1 probably upregulates beta oxidation making the cell fat adapted when the C16 appears, and the fact (seeing as the cells aren't actually "working" because no nervous system, and there's glucose or excess glucose and insulin present) that C18:1 oxidation can be uncoupled more really helps with this fat adaptation.
3) the more fat you burn, the less desirable uncoupling is, unless you're Inuit in native land. On an endurance run, you'd certainly want the least uncoupled energy source in your mitochondria. This would be a good reason for the bulk delivery system (lipoproteins) to prioritize SFA loading and offloading in the fat burning state.
That mostly fits in with these:
https://www.ncbi.nlm.nih.gov/pubmed/12177411
https://www.ncbi.nlm.nih.gov/pubmed/17369521
If you can't form oleate you can't store fat so you burn it, uncoupling if necessary. If you do oxidise pure palmitate, certainly in in beta cells, it's bad news. I'd guess the few survivors are using any oleate from the diet...
Peter
That's consistent with the ob/ob mice getting a buildup of ceramide in their pancreases instead of just burning the palmitate (I assume they eat more than the lean mice). I basically see ceramide as gunk, a waxy endproduct easier to make than to remove. Regardless of any special reactive properties, excess ceramide is going to stop membranes being good mobile bases for proteins, bringing down metabolism.
This is probably a wrong view, but it works visually for me.
Someone reminded me that, while the thyroid hormone T3 lowers LDL, rT3 binds to the LDL receptor and downregulates it. Now why would that be? Why would a hormone variant you make when burning fat instead of sugar act to keep ex-VLDL lipoproteins in circulation for longer so as many TGs as possible can be extracted from the bottom of the barrel? Could it be for the same reason that dietary saturated fats downregulate this receptor, in the circumstances in which this happens?
this mechanism could be useful if it's general:
Low density lipoprotein delays clearance of triglyceride-rich lipoprotein by human subcutaneous adipose tissue
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3622338/
Post a Comment