You sometimes have interesting conversations in reception. A lady commented to me that she had no trouble getting arthritis meds in to her dog, some difficulty with antibiotics and that there was no way that she could persuade him to take the tramadol which had been prescribed as a supplementary analgesic for his arthritis.
I mentioned that he might not find the psychotropic effects of tramadol pleasant. A bit like morphine, not everyone finds the effects enjoyable. She concurred about the effects of morphine. It turned out that she had once been given morphine after surgery and had lost her automatic ability to breathe.... A sort of iatrogenic and thankfully temporary Ondine's curse. More poetically here.
In her case the solution had been that a nurse was assigned to sit by her bed and remind her to take a breath whenever she forgot, until automaticity of breathing returned as the morphine wore off. It's a rare side effect.
It started me thinking about control systems. The pancreas monitors blood glucose and responds with insulin and glucagon. It has a generous nerve supply with a whole host of neurotransmitters affecting insulin output. There are also glucoreceptors in the aortic bodies feeding information to the autonomic nervous system. There are, not surprisingly, nuclei in the brain which have glucosensors. There are probably insulin receptors in the brain too.
I think it is just worth pointing out that I have absolutely no problem with the fact that the brain controls blood glucose. Not the pancreas, not the liver, not the adipocytes. Long term stability needs brain input. You could probably say the same about body weight.
It's an integrated system.
But I doubt very much that the brain is central to the bulk management of metabolism. You would hardly expect a regulatory system such as the pancreas/liver/glucose axis to be intrinsically unstable and only kept functional by continuous and aggressive brain intervention. Metabolism ought to be largely self regulatory, with fine tuning by the brain to meet specific conditions.
Aside: Apparently the Eurofighter is intrinsically unstable in flight and is kept stable by enormous computing power and continuous hardware activity. It uses this intrinsic instability, given tightly controlled expression, in order to flip the aircraft from zero to 7 G in a completely unreasonable period of time. Keeping the pilot conscious under these conditions is the challenge to be met. I only heard any of this because we had an interesting presentation about this latter aspect at an anaesthesia meeting on cerebral perfusion pressures not so long back... Back to metabolism:
There is clearly a regulatory set point for the control of breathing. There is also one for blood pressure, blood sodium, potassium, pretty well everything else. I manipulate many of these daily to earn a living. Why not one for bodyweight?
At the moment I have an insulinocentric view of metabolism and bodyweight. Insulin appears to explain a fairly large chunk of weight control issues. It doesn't intrinsically need a set point concept, but there is every reason to accept some brain input to determine bodyweight. But the idea that the brain can over ride the obesogenic effect of a diet which requires chronic hyperinsulinaemia to maintain a semblance of health is very hard to accept.
It was working through these concepts that I dug out a fascinating paper on leptin and blood glucose emailed to me by Stephan a little while ago. I finally got to read it in depth last week.
The streptozotocotin diabetic rat strikes me as a pretty good model for type 1 diabetes. It looks to be a pure beta cell failure model, ie it lacks the stupidity of considering metabolic freaks such as the Otsuka diabetic rat as in any way representative of any form of human type 2 diabetes. I have time for STZ diabetic rats as being a model for type 1 diabetes. STZ produces acute, persistent and profound hypoinsulinaemia.
The researchers made some rats severely diabetic with STZ (BG > 400mg/dl), then infused leptin in to the cerebrospinal fluid within the third ventricle of the brain.
End result: Normoglycaemia. No change in blood insulin level.
That's quite impressive. No, it's bloody amazing. They did a ton of other things too and had a number of excellent control groups. The conclusion the researchers reached is that there appears to be neural control of hepatic glucose synthesis and output, plus neural control of muscle glucose uptake. They're probably correct. It's a very interesting paper. But is a massive intracranial infusion of leptin genuine physiology or is it pushing a fine tuning system to its ultimate limits using a chemical gorilla? Turbocharging a Morris Minor to out-accelerate a performance BMW?
They were putting almost as much leptin per minute in to the CSF/brain volume as would be produced by whole body fat mass. They probably achieved intracerebral concentrations of leptin way above what might every be produced in plasma.
Of course the goal of the group is to produce a centrally acting leptin mimetic to treat diabetes. Hmmmm, the unintended consequences will be interesting!
As always, picking through the results tables provides a wealth of information to be fitted in to the insulinocentric view of metabolism.
Let's look at the blood insulin levels:

It looks like STZ produces profound hypoinsulinaemia. It doesn't matter what else you do, insulin is low after STZ. No problems here. Hypoinsulinaemia across the board.
Before we look at blood glucose levels let's peek at leptin levels:

Interesting? Well the rats in the control non-diabetic group have lots of leptin. Unless the rat is being given a subcutaneous infusion of leptin (right hand column) there is very little leptin in the blood stream of the diabetic rats. Why so? STZ does nothing to leptin production. But hypoinsulinaemia does a great deal to lipolysis. These rats have had uncontrolled type one diabetes for a couple of weeks before the six day experiment. We don't get any bodyweights in the paper but leptin reflects adipose tissue mass. The STZ rats are profoundly fat depleted compared to the non diabetic controls. I think they're emaciated. This thin:
"WAT [White Adipose Tissue] had clearly atrophied in size in STZ-SAL rats. WAT in STZ-LEP and STZ-PF rats was further atrophied compared with the STZ-SAL group. For that reason, we could not extract enough RNA for Northern blotting"
That's thin.
So now let's look at weight change. During the 6 day experiment the non diabetic control rats gained about 15g. The hypoinsulinaemic groups all lost 15-25g bodyweight (except the semi starved group in column 4, starvation of type one diabetics is not advised, ND for leptin in the above graph wasn't that they didn't look for leptin, it's just the amounts present where below the limits of their assay).
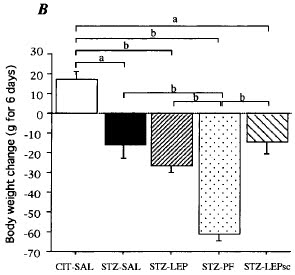
This is irrespective of whether they were spilling glucose through their kidneys or not. Hypoinsulinaemia allows lipolysis. Drop insulin and you drop fat, however much rat carbohydrate-crap-in-a-bag you eat. Intracerebral leptin does not stop this.
Now let's look at food intake:

STZ diabetic rats become hyperphagic. At least when they are fed on rodent chow and allowed free access. High carbohydrate, low fat. As the authors point out this does not happen if you feed a high fat diet but I've blogged about this elsewhere.
I'd just like to consider why this hyperphagia should occur from the hang-glider perspective, rather than the Eurofighter perspective.
STZ rats lack insulin. They do lipolysis to extreme levels and run their muscles on fatty acids. Until there is not a lot of fat left, as reflected by hypoleptinaemia. Down on the ground the cells needs calories but there is not enough fat left for this level of hypoinsulinaemia to provide free fatty acids for metabolism. But it is possible to get blood glucose levels high enough to use non GLUT4 transporters to get glucose into muscle cells (see here). With hyperglycaemia there is a chance of survival. To do this you need marked hyperglycaemia. You need it, so metabolism does its best to provide it. In fact these rats will have been hyperglucagonaemic to help raise their blood glucose.
So the rats will eat a ton of glucose precursor, fail to retain it in the liver due to hypoinsulinaemia, add as much to it, using glucagon, as possible and push glucose in to muscles using the brute force of a concentration gradient. The cost is calorie loss through glycosuria and whole body hyperglycaemic damage. But death is postponed. Life is tenaceous.
The STZ control rats ate over twice as much crapinabag as the non STZ controls while continuing to lose weight hand over fist.... Good old hypoinsulinaemia and the body's adaptation to it.
The paper certainly gives support to the concept that the brain is significantly involved in glucose homeostasis. How this will relate a fat mass set point is a line of thought I'm looking forward to seeing Stephan develop.
I rather like this paper. It doesn't need me to move from my insulinocentric bias for weight loss. Ah, good.
Back to LC for diabetes and adaptation to high carb next.
Peter

8 comments:
http://www.ncbi.nlm.nih.gov/pubmed/21136047
*K2 prevents hyperglycemia in STZ-D rats...nothing about insulin in the abstract though
Hi Peter,
Nice to see you back to blogging.
Basal insulin signaling is required to restrain lipolysis, so you get uncontrolled fat loss in type 1 diabetes both in animals and humans. With no fat, you also have very little leptin, so uncontrolled T1DM is really a lack of insulin and leptin.
The reason those rats were hyperphagic yet couldn't put on weight is because they were peeing out a huge amount of glucose. If you subtract the calories lost in urine from the total energy intake, their energy balance makes sense.
The fact that a basal amount of insulin signaling is required to maintain fat mass does not imply that postprandial, carb-induced insulin spikes promote fat gain in metabolically normal individuals.
The role of the leptin body fat homeostasis system is to set the "gain" on all the other processes that influence body fatness. For example, when leptin-sensitive neurons are activated, they inhibit food reward pathways and stimulate the pituitary into a state that signals bodywide fuel repletion. Those systems mostly function independently of leptin, but are modulated by it.
http://www.sciencedaily.com/releases/2011/01/110126161835.htm
These findings in mice show that insulin becomes completely superfluous and its absence does not cause diabetes or any other abnormality when the actions of glucagon are suppressed
Hi John,
OK, so what exactly is going on there????
Hi Stephan, yes, I can see that the math would stack up to balance the equation. I'm still wondering about the physiological benefits of hyperglycaemia in the face of hypoinsulinaemia, with all of the attendant costs. Can't really see how an evolutionary pressure would work this way but it's an interesting thought train.
I can certainly see that there must be some sort of set point for fat. Sticking a large respiratory dead space in to an anaesthetic circuit gives an uncorrectable elevation in arterial CO2 if the patient is breathing spontaneously. I'm thinking along the lines of fasting hyperinsulinaemia messing with the fat set point in the same way.
Reducing fasting insulin would allow weight to drop to some preferred set point and would show as a decrease in spontaneous food intake..... I would agree that post prandial hyperinsulinaemia is far less significant.
I know Chris M pointed out that the (fasting) hyperinsulinaemia following portosystemic diversion of pancreatic drainage does not cause obesity but I can't find any long term studies with the weight changes documented (yet) to support or deny this..... Still looking.
Anyway, lots of thinking to do
Hi stormrunner,
Interesting. I spent some time looking at the glucagon response to ingested protein as a counterbalance to its insulinotropic effects but the levels induced appear to be too low to trigger any sort of lipolysis. Now the marked hyperglucagonaemia of diabetes may be a different matter.....
The main problem with this approach is that total pancreatectomy which should abolished both insulin and glucagon (and a few other hormones) in dogs and humans seems to produce hyperglycaemia.
There is a possibility that total pancreatectomy in herbivores may be relatively harmless but, although I've looked for publications on this, I've yet to find one.
Peter
Hi Peter,
All I have read are scattered trials with K1 or K2 showing some degree of increased insulin sensitivity or glucose tolerance, which of course doesn't really explain the K2 effect in STZ-Ds. This* shows K2 actually increasing the STZ-induced decrease of fasting insulin. Searching pubmed for "K2 leptin" doesn't give anything...
*http://www.ncbi.nlm.nih.gov/pubmed/11214355
Are the dogs and humans subjected to pancreatectomies getting hyperglycemia on high fat?
@Peter:
You said " I spent some time looking at the glucagon response to ingested protein as a counterbalance to its insulinotropic effects but the levels induced appear to be too low to trigger any sort of lipolysis. Now the marked hyperglucagonaemia of diabetes may be a different matter....."
I think the key is that hyperinsulinemia by itself is not bad. Insulin is antinflammatory. Glucose is the main stimuli for insulin secretion because hyperglycemia is pro-inflammatory. I havent looked at any reference for amylin secretion patterns...maybe most amylin is secreted during first-phase insulin secretion?
Good to be participating in such discussions...really helps to know the medications and the after effects of the medications in individuals.
Medical Billing Software
Using leptin to inhibit glucagon in an intact pancreas wouldn't be the same as with the total lack of a pancreas, though. The brain might receive some signal from the pancreas as a result of the leptin, or the leptin might encourage or discourage the pancreatic secretion of who-knows-what-else that might affect blood glucose regulation. Or in the total absence of glucagon, some other, less subtle regulator of glucose might become dominant?
I guess we couldn't expect much endogenous leptin production after a pancreatectomy.
Centrally administered leptin is very bad for bone mass. Leptin shows up in animals with skeletons.
Post a Comment