Gotcha Eric.
Sunday, April 03, 2022
Monday, March 28, 2022
Linoleic acid panel discussion
Here's the link to the panel discussion about PUFA and obesity, hosted by David Gornoski.
My Big Fat Panel: How Seed Oils Cause Obesity - A Neighbor's Choice by David Gornoski
It was an interesting discussion but I'm not really sure we achieved any consensus as to how you would convince a mainstream scientist that we are correct...
Peter
Wednesday, March 16, 2022
Friday, February 18, 2022
Ioannidis
I have great respect John Ioannidis
I'm also coming to accept that prison or sectioning for the primary malfeasants is not going to be the best or even a practical solution. Lessons still have to be learned and systems questioned.
I'm also coming to accept that prison or sectioning for the primary malfeasants is not going to be the best or even a practical solution. Lessons still have to be learned and systems questioned.
Peter
Monday, January 31, 2022
How's it going, Pfi$rael?
There may be some readers who genuinely believe the pfvacine really does protect agains severe illness and even death from covid. That's fine by me.
Using the crudest of datasets it looks like Israel's omicron wave peaked on January 26th 2022 by positive test result.
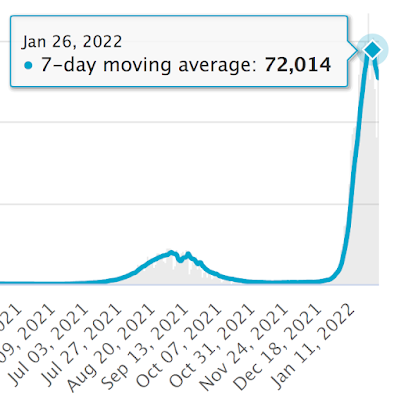
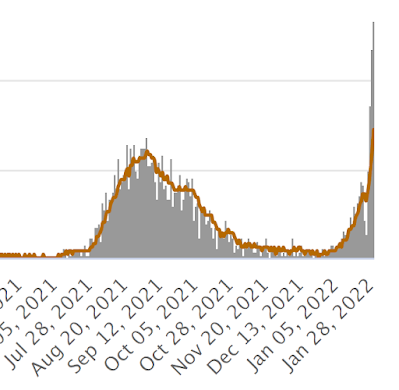
Quite how long the delay will be for the death toll to peak is a little hard to guess with omicron, but if it's three weeks then the hypervaccinated Israelis are in for a hard time over the next two weeks:
It's worth bearing in mind that it is currently almost impossible to die in a hospital in the UK without testing positive for SARS-CoV-2. I presume Israel is the same, so some of these fatalities may be incidentally positive test results, in which case the mortality peak will follow sooner than expected after that of "cases".
I have an elderly relative approaching end of life care for cancer who was hospitalised about a week ago and tested positive four days after admission. Incidentally she is unvaccinated and completely asymptomatic. Sadly she has now fallen and broken her hip, with an urgent repair planned for about now.
She is, undoubtedly, a covid hospitalisation statistic. The likely outcome seems uncertain.
Peter
Friday, January 28, 2022
Saturday, January 22, 2022
So you want some DHA? Chickens in Norway
Again via twitter, Tucker retweeted a thread which included the link to this paper
Increased EPA levels in serum phospholipids of humans after four weeks daily ingestion of one portion chicken fed linseed and rapeseed oil

which comes from another paper:
It shows, very clearly, that in rats both linoleic acid and alpha linolenic acids are equally efficacious at suppressing the conversion of ALA to DHA. Given 1% of energy in the diet as ALA then copious amounts of DHA are produced, just so long as the LA proportion of calories is less than 2% of the total. Never mind the ratio. Avoiding PUFA will generate DHA from ALA provided a basic minute minimum of ALA is present in the diet, somewhere around 1% of calories. Over at the right hand side of the plot we can see that even drinking 12% of your energy intake as ALA will not generate significant DHA if you are up in the cardiological nirvana of 16% of energy as LA.
That is in rats.
Chickens superficially appear to be somewhat different.
Switching from soybean oil to a rapeseed/linseed oil mix in the diet increases DHA in the breast meat. Soybean oil gives 14% of lipid as DHA, rapeseed/linseed gives 21%. Of not very much fat in breast meat so the amounts are small in total, but still highly statistically significant.
So ALA in food bumps up DHA in muscle. Of chickens.
This is what the chicken diet looked like, with annotation to give the fat percentage of total calories and the LA and ALA percentages of total calories in the diet, crudely:
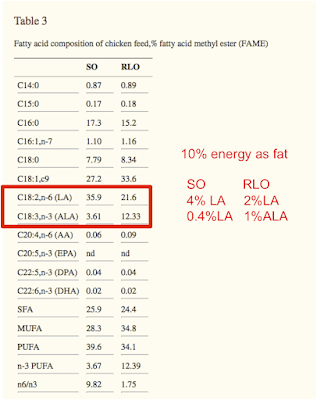
We can overlay the chicken diet very approximately, on the rat plot to get this:
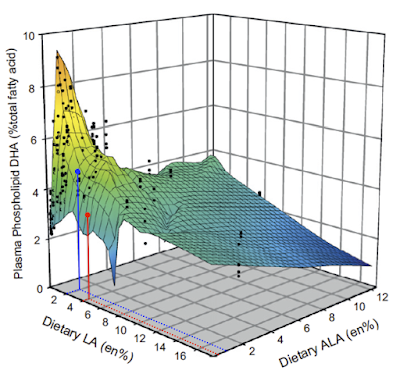
which, not surprisingly, suggests that the DHA production in the "high" ALA diet (blue) is actually more influenced by the reduction in LA. At 4% LA we could equally have had ALA at 1% of calories and still got a lower value for DHA than we did by having LA down at 2% of calories.
Might this work for humans too? Could adding ALA to our diet improve DHA availability, with all of what that entails for improved brain development and cognitive function. Just by avoiding LA and maybe drinking a little (traditional) varnish? The study didn't ask this. Instead they fed the above chicken to some humans. Either omega 3 enriched or omega 6 enriched.
This actually dropped DHA in the participants' plasma phospholipids in both groups. Admittedly not by much, so the change was neither statistically nor biologically significant. But it dropped. I would hazard a guess that the chicken simply displaced a richer source of ready-formed DHA from the diet, the tiny amount in the breast meat would do nothing per se. I believe Norwegians consume a certain amount of fish, unless they're given free chicken to eat.
The rise in EPA must have made the authors happy that something positive came from all of that work. But I don't think you can build a brain out of EPA, DHA looks to be the molecule for that.
Ultimately pre-formed DHA does not look to be necessary for brain development if you have a modest supply of ALA from (grass fed, possibly large) animals and avoid consuming significant amounts of LA. This appears to hold true for rats, chickens and I expect for humans.
Peter
Thursday, January 13, 2022
Covid playground
This is just a "post" to allow comments between people interested in the current COVID saga more easily. Comments on all posts older than two weeks are to be moderated by myself, just to keep the spam under control. So here are two weeks of unmoderated commenting scope if people want to exchange comments if I'm off working or weekending with the kids.
There you go Eric. Good idea.
Peter
Wednesday, January 12, 2022
Rimonabant and adipocytes
Okay. I hope everyone has heard this podcast with Raphi chatting to Tucker and Amber:
CB1 agonists make adipocytes insulin sensitive. Rimonabant blocks this effect, which made me feel good about Protons/insulin/obesity and I left the subject alone in a nice glow of confirmation bias for a few months.
Nothing changes in the gut. Nothing changes in the liver. Nothing changes in the brain. The hypothalamic Reward™ dopaminergic neurons are left untouched. All that happens is that adipocytes lose (at least) the insulin sensitising effect of CB1 receptor activation. Weight completely normalises in less than a month. It's also worth noting that in control mice adipocyte CB1 gene deletion does nothing.

I feel Amber articulated the adipo-centric view rather well. The whole podcast reminded me of a few ideas I've had kicking around for ages and it might now be time to do a bit more posting.
I was taken back to good old Rimonabant by some of Tucker's points. It got a passing mention in an old blog post back in 2010. I may not have been terribly impressed at the time.
Rimonabant and hemopressin
For those of us with an adipocentric/insulin based outlook on life Rimonabant is interesting. It takes about 30 seconds on PubMed to pull out
Rimonabant and hemopressin
For those of us with an adipocentric/insulin based outlook on life Rimonabant is interesting. It takes about 30 seconds on PubMed to pull out
CB1 agonists make adipocytes insulin sensitive. Rimonabant blocks this effect, which made me feel good about Protons/insulin/obesity and I left the subject alone in a nice glow of confirmation bias for a few months.
But these are isolated cells, does anything like this happen in real life? I would expect Rimonabant -> adipocytes -> reduced insulin signalling -> release of free fatty acids -> weight loss -> reduced appetite.
In that order.
Central to this is that the brain senses energy availability (Amber cited work I'd never heard of, my own ideas are just ideas. I felt it was self evident. You could say I just made it up) and reduces appetite when energy availability is good. Rimonabant frees up calories from adipocytes. But it is nasty stuff in the brain.
Developing drugs which do not pass the blood brain barrier is old hat in anaesthesia. Doing the same for CB1 receptor blockers seems pretty simple too. Just imagine, all the weight loss, none of the suicidal ideation. There are several under development.
But peripheral CB1 blocking drugs hit all sorts of targets ranging from the gut through the liver to the vagus nerve. And adipocytes.
What would an adipo-centrist look for?
Obviously we want an adipocyte specific CB1 receptor knockout mouse. It just has to dawn on you that that is what you want. Which took a while in my case.
So another 30 seconds on PubMed gave me this one:
Adipocyte cannabinoid CB1 receptor deficiency alleviates high fat diet induced memory deficit, depressive-like behavior, neuroinflammation and impairment in adult neurogenesis
and a little wander to the Place-which-shall-not-be-named gets you the full text.
Adipocyte cannabinoid CB1 receptor deficiency alleviates high fat diet induced memory deficit, depressive-like behavior, neuroinflammation and impairment in adult neurogenesis
and a little wander to the Place-which-shall-not-be-named gets you the full text.
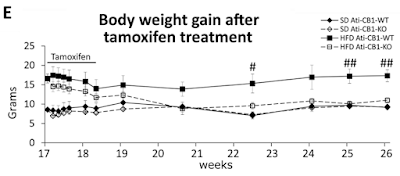
They built a mouse model with a tamoxifen trigger-able deletion of the CB1 gene, specifically in adipocytes. How the hell they do that I can't follow but it's in the methods with links (not followed by me this time I'm afraid). I just have to accept that they can do it and that the technique is very, very clever. Then they fed a German high fat, high linoleic acid diet (around 10% calories from LA, ballpark) to make the mice fat over several months, leading to the start point at week 17 of graph E below. Then they injected tamoxifen daily for 10 days to induce permanent deletion of the CB1 receptor gene in adipocytes only. Here's what happened to the weights.
Black squares are fat mice which keep their CB1 receptors. Open squares are fat mice which lose them. D Both diamonds are controls:
Nothing changes in the gut. Nothing changes in the liver. Nothing changes in the brain. The hypothalamic Reward™ dopaminergic neurons are left untouched. All that happens is that adipocytes lose (at least) the insulin sensitising effect of CB1 receptor activation. Weight completely normalises in less than a month. It's also worth noting that in control mice adipocyte CB1 gene deletion does nothing.
Ultimately the adipo-centric view has phenomenal explanatory power when backed up by the insulin ROS concept. Everything else is higher level signalling and unexciting to me.
There are other interesting things in the paper relating to food intake and uncoupling (ie the rapid weight normalisation occurred without reduction in food intake) but I'll call it a day for now. Except to mention that in human victims of Rimonabant it is well recognised weight loss is greater than can be accounted for by reduction in food intake. Fascinating.
At some time I'll drag myself back to working out the F:N ratio of mixed fats. Two different butters, two lards, one plus 5% soybean oil, done so far. Still got coconut oil and fully hydrogenated coconut oil to go. Losing the will to live.
Perhaps I should ask my son to write me a bit of software to do this!
Peter
Saturday, December 11, 2021
Protons (67) a formula revised for butter oil
A couple of months ago Tucker emailed me this study
Docosahexaenoic acid lowers cardiac mitochondrial enzyme activity by replacing linoleic acid in the phospholipidome

Docosahexaenoic acid lowers cardiac mitochondrial enzyme activity by replacing linoleic acid in the phospholipidome
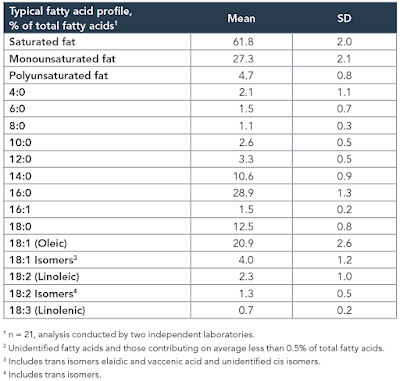
The study itself, while interesting, is not the point. The point is that the diet used contained the infamous 42% of calories from fat and was obesogenic. In common with a number of other studies I have been mulling over, the fat was butter oil. This is the butter oil composition
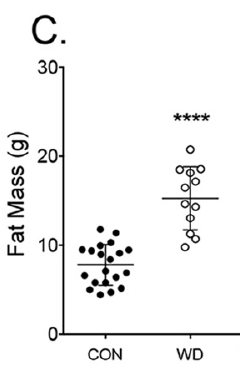
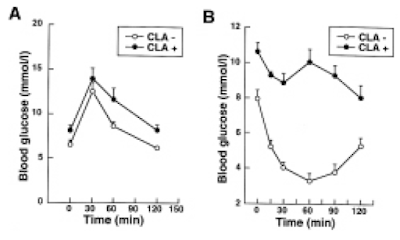
which has 2.3% linoleic acid in 42% of calories giving just over 1% of calories as LA. All I am interested in here is the comparison between 14 weeks on control low fat diet (CD) vs 14 weeks on a low linoleic acid Western Diet (WD). Here are the fat mass data (bodyweight mirrored this)
and here are the IPGTT results. We're just comparing the black control (CON) with the red western diet (WD) lines
 |
So I think we can say that 14 weeks on butter oil is obesogenic and has the appropriate insulin resistance as we expect from any obesity with its increased basal lipolysis of large adipocytes coupled with Protons effects on the non adipose tissues.
Aside:
The diet looks like this:
If anyone thinks this looks like fudge, here is the recipe for a kilo of the stuff
Take 340g of table sugar, add 200g of anhydrous butter fat, bulk it up with 150g of corn starch to help it set and throw in some casein if you feel like it. Mix and extrude (you could cut in in to cubes for human consumption).
Fudge.
Looks yummy. Rewarding. Don't snigger!
End aside.
Butter oil is one of those features of study designs which produce obesity without linoleic acid.
Again, prompted by Tucker, I re-drafted the F:N ratios of MUFA and PUFA in the last post to account for the consumption of one NADH to provide an NADPH for rearranging each double bond during beta oxidation and we can add these revised ratios and some for selected saturate values to the composition table of butter oil like this:
In red are linoleic acid and the short chain fatty acids of an equal or lower F:N ratio cf LA. I threw in oleic acid and C8 caprylic (blue numbers) too to point out that caprylic acid, though higher than LA, is now lower than oleic (I view oleic acid as the mammalian default for "normal" insulin sensitivity) and so might be obesogenic.
So, from the F:N ratio Protons perspective, we have a modest supply of short chain fatty acids of potentially greater insulin sensitising ability (hence obesity promoting) than linoleic acid itself.
The effect of butyrate as a dietary supplement on obesity is controversial and reviewed here
Butyrate: A Double-Edged Sword for Health?
Conclusions totally depend on how you set your study up, what you consider good and what you consider bad. Bear in mind that butyrate is the darling of fibre-philes so consider publication bias too. Conversely it is to a large extent consumed by the colonic epithelium, so not a lot gets through to the systemic circulation. But some clearly does. The snippet of Figure 2 which caught my eye was this section:
Butyrate: A Double-Edged Sword for Health?
Conclusions totally depend on how you set your study up, what you consider good and what you consider bad. Bear in mind that butyrate is the darling of fibre-philes so consider publication bias too. Conversely it is to a large extent consumed by the colonic epithelium, so not a lot gets through to the systemic circulation. But some clearly does. The snippet of Figure 2 which caught my eye was this section:
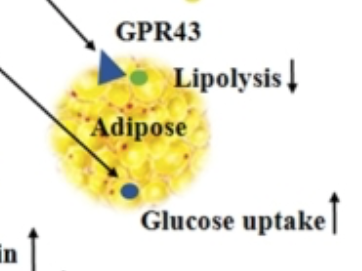
I would just point out that anything which decreases lipolysis and increases adipocyte glucose uptake is NOT going to make you skinny. It might make you insulin sensitive (bravo), at least until you get fat enough to leak excess FFAs via augmented basal lipolysis.
You could of course just say butter oil fudge is highly Rewarding, so makes rats and mice fat. How can you tell it's Rewarding? Because it makes rats and mice fat. Except corn oil is also very Rewarding but ad-lib preferential consumption fails to induce obesity. For obesity there is the absolute necessity of calories to enter adipocytes and then stay there. Long term. Dopamine release in the brain might make you choose to eat something over something else but without pathological energy storage... Shrug.
My own concept of how butter oil/sucrose causes obesity is limited by the clear fact that there is no way of simply saying a given F:N ratio will always produce obesity. Too many variables for this to be set in concrete, which allows the hypothesis to side step conflicting evidence. You have been warned.
Random thought: Sucrose (when it doesn't produce a slim insulin sensitive phenotype) usually produces hyperinsulinaemia and insulin resistance (often "skinny fat"). The higher the insulin levels the more effective insulin sensitising dietary components (linoleic acid and now possibly SCFAs) are at allowing that high level of insulin to generate obesity. Probably why the cornstarch is added to the fudge, to augment the insulin response.
As always, alternative explanations welcome.
My thanks to Basti in the comments after the last post which crystalised a lot of this current post. And to Tucker twice over.
Peter
Wednesday, December 08, 2021
Protons (53) a formula revised
Back in Protons (53) a formula I wrote down how to work out the F:N ratio of (even chain) fatty acids with varying double bonds:
where n is the length of the carbon chain and db is the number of double bonds.
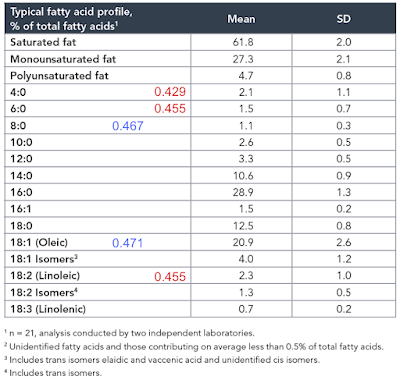
Oleic C18 is 18-1-1 divided by 36-1, ie 16/35 = 0.457
Linoleic 18-1-2 divided by 36-1, ie 15/35 = 0.423
This is fine up to C18 but C20 and above are targeted to peroxisomes rather than mitochondria so the need for an F:N ratio fades. Peroxisomes have their own signalling systems but research on them is in its infancy.
Anyhoo, Tucker mentioned off blog that during the multistep processing of double bonds there is a step which consumes NADPH. This will have to be re-reduced from the resultant NADP+ by the Krebs Cycle where NADH producing steps have iso enzymes capable of generating NADPH instead of NADH. That reduces the NADH supply to the electron transport chain by 1 NADH per double bond requiring NADPH, so complicates the formula.
The formula ends up as:
F/N = (n-1-db)/(2n-1-db)
It makes a relatively small change to the ratio as the denominator is a much larger number than the numerator.
Oleic acid, originally 0.457 becomes
18-1-1 divided by 36-1-1, ie 16/34 = 0.471
and linoleic acid, originally 0.423 becomes
18-1-2 divided by 36-1-2, ie 15/33 = 0.455
The latter is interesting as it moves linoleic acid upwards towards MUFA and the saturates because the denominator drops.
The value for caprylic (shortest saturate in common consumption) is 0.467 and with the new LA now at 0.455, they are getting closer. Also caprylic is now at lower F:N ratio than oleic. I just wonder if this is part of the explanation of the coconut based diets used by Surwit to induce obesity with LA still limited to 4% of calories...
Thanks to Tucker for the NADPH requirement insight.
Peter
Tuesday, December 07, 2021
!Kung Bushmen and mongongo nuts yet again
Well, I got that wrong about conjugated linoleic acid (CLA) from mongongo nuts.
The !Kung people eat their mongongo nuts and the large amount of alpha-eleostearic acid converts to 9cis, 11trans CLA:
Alpha-eleostearic acid (9Z11E13E-18:3) is quickly converted to conjugated linoleic acid (9Z11E-18:2) in rats
This 9c, 11t CLA is exactly the same isomer as rumenic acid, the primary CLA of ruminant meat/dairy fats.
Alpha-eleostearic acid (9Z11E13E-18:3) is quickly converted to conjugated linoleic acid (9Z11E-18:2) in rats
This 9c, 11t CLA is exactly the same isomer as rumenic acid, the primary CLA of ruminant meat/dairy fats.
It's not a lipolytic agent. Not from monongo nuts, not from ruminants.
For lipolysis you want 10trans, 12cis CLA.
Manufacturing a bulk supply of CLA for marketing as a fat loss drug uses a process of treating ordinary linoleic acid with a catalytic industrial process which isomerises the LA in to roughly a 50:50 mix of 9c, 11t CLA and 10t, 12c CLA plus some odds and sods
The 10t, 12c isomer is a lipolytic agent of some potency. There's a nice review here:
However it does not appear to be found as a normal component of any biological system as far as I know, though I'm open to someone finding a source. At the moment it looks like it is a drug, manufactured from linoleic acid by an industrial process. The chemical formula might be identical to rumenic acid but on a "shape", charge distribution and metabolism basis (the location and orientation of the double bonds really matters to enzymes) the two have nothing what so ever in common.
In addition to weight loss 10t, 12c CLA can also trigger adipocyte apoptosis. A little apoptosis might be fine if you have adipose tissue hyperplasia (too many adipocytes, rather than too distended adipocytes) but if you have normal levels of overly large adipocytes it will place the burden of accepting excess insulin mediated lipid for storage in to the remaining, already overly large, adipocytes. Or your liver.
This is essentially a lipodystrophy, certainly if taken far enough. As in congenital and acquired lipodystrophies, this will be associated with glucose intolerance, insulin resistance and functional type 2 diabetes. In rodent models you can drive this process somewhat further than you can in human clinical trials. So this study uses mice:
Conjugated Linoleic Acid Supplementation Reduces Adipose Tissue by Apoptosis and Develops Lipodystrophy in Mice
Bear in mind this is a model and has been set up to produce an extreme black/white result and it delivers.
Conjugated Linoleic Acid Supplementation Reduces Adipose Tissue by Apoptosis and Develops Lipodystrophy in Mice
Bear in mind this is a model and has been set up to produce an extreme black/white result and it delivers.
Oral glucose tolerance test and intraperitoneal insulin tolerance tests:
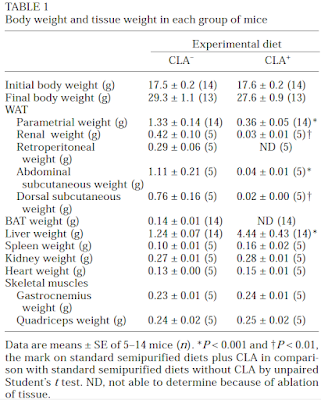
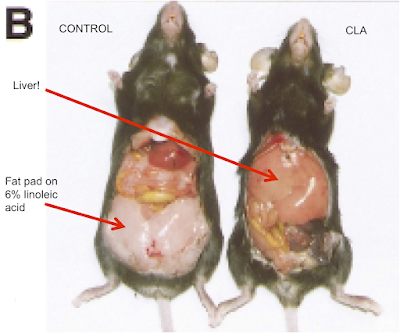
Here are the weights of various organs of interest (check the liver):
Note that the model was set up so there was no weight loss with the 10t, 12c CLA treated group. There is massive adipose tissue loss, adipocyte number loss but no weight loss (it's a model, people are clever). All of the lipid which should be in adipocytes ends up in the liver. What does a 4.44g liver look like in an adipocyte depleted mouse? Like this:
This is the diet:
"The semipurified diet was a low-fat diet and on a calorie basis contained 63% carbohydrate, 11% safflower oil, and 26% protein. Safflower oil was used as a source of fat. Safflower oil (high-oleic type) contained 46% oleic acid (18:1 n-9) and 45% linoleic acid (18:2 n-6) from total fatty acids. CLA was prepared as a free fatty acid at Rinoru Oil Mills (Nagoya, Japan) and stored frozen in plastic bottles blanketed with nitrogen. Linoleic acid was isomerized to CLA with isomers (34% c9, t11/t9, and c11; 36% t10 and c12; 3% c9, c11/c10, and c12; 2% t9, t11/t10, and t12 from total fatty acids). In the CLA-fed group, to keep fat intake constant in the 2 groups, 25% of the safflower oil was replaced with CLA"
That's a quarter of 11% of calories as mixed isomer CLA, the sort you might take as a supplement, ie around 3% of calories. About a third of this is the active 10t, 12c CLA, ie around one percent of calories.
Comparison of dietary conjugated linoleic acid with safflower oil on body composition in obese postmenopausal women with type 2 diabetes mellitus
they were using 6.4g/d mixed CLA isomers. That will be around 3g/d 10t, 12c CLA. That's exactly the ball park used to produce lipodystrophy and diabetes in mice. The same phenomenon occurs in pigs where after slaughter back fat can be extracted, weighted and processed to detect apoptosis:
Supplementation with conjugated linoeic acid decreases pig back fat deposition by inducing adipocyte apoptosis
"The semipurified diet was a low-fat diet and on a calorie basis contained 63% carbohydrate, 11% safflower oil, and 26% protein. Safflower oil was used as a source of fat. Safflower oil (high-oleic type) contained 46% oleic acid (18:1 n-9) and 45% linoleic acid (18:2 n-6) from total fatty acids. CLA was prepared as a free fatty acid at Rinoru Oil Mills (Nagoya, Japan) and stored frozen in plastic bottles blanketed with nitrogen. Linoleic acid was isomerized to CLA with isomers (34% c9, t11/t9, and c11; 36% t10 and c12; 3% c9, c11/c10, and c12; 2% t9, t11/t10, and t12 from total fatty acids). In the CLA-fed group, to keep fat intake constant in the 2 groups, 25% of the safflower oil was replaced with CLA"
That's a quarter of 11% of calories as mixed isomer CLA, the sort you might take as a supplement, ie around 3% of calories. About a third of this is the active 10t, 12c CLA, ie around one percent of calories.
If a human consumes 2000kcal/d then that's 20kcal or 2g of 10t, 12c CLA per day. In this now rather well thumbed study
Comparison of dietary conjugated linoleic acid with safflower oil on body composition in obese postmenopausal women with type 2 diabetes mellitus
they were using 6.4g/d mixed CLA isomers. That will be around 3g/d 10t, 12c CLA. That's exactly the ball park used to produce lipodystrophy and diabetes in mice. The same phenomenon occurs in pigs where after slaughter back fat can be extracted, weighted and processed to detect apoptosis:
Supplementation with conjugated linoeic acid decreases pig back fat deposition by inducing adipocyte apoptosis
Comparable studies would be difficult in humans but least pigs aren't mice.
Where does this leave the !Kung and their mongongo nuts? Well, they certainly never see any 10t, 12c CLA, our liver only converts alpha-eleostearic acid to rumenic acid (assuming we're like rats). This latter is either a weak or non lipolytic/apoptosis agent. Does that leave the !Kung as inexplicable?
No.
It turns out that alpha-eleostearic acid is a rather potent lipolytic agent in its own right, it also induces apoptosis in fat cells in a similar manner to 10t, 12c CLA. Bitter melon seed oil is another, quite well studied source of alpha-eleostearic acid. This gives the flavour:
Mongongo nuts are lipolytic until their alpha-eleostearic acid content is detoxified to rumenic acid. To me, this suggests that living on mongongo nuts may carry weight control benefits at some risk of generating a degree of lipodystrophy, however small. I doubt anyone has gone studying adipocytes from the !Kung for markers of apoptosis. It looks like there will be a trade off between degree of lipolysis, giving small, low basal lipolysis adipocytes vs lost adipocytes giving larger, more basally lipolytic remaining adipocytes. I suspect the dose makes the poison.
I think it's probably unimportant to go in to detail about how alpha-eleostearic acid and 10t, 12c CLA induce lipolysis/apoptosis but, not surprisingly, it involves the generation of ROS for both.
Peter
Monday, November 29, 2021
Are you on clenbuterol? (3)
More from Risérus
Trans fatty acids and insulin resistance
"This is especially true [inducing insulin resistance] for conjugated TFA, i.e. conjugated linoleic acid (CLA), which clearly impairs insulin sensitivity."
I think is reasonable to assume that Risérus expects ordinary trans fatty acids to impair insulin sensitivity too, though not quite as effectively as CLA does. He just needs a big enough intervention study to prove it.
Of course he is wrong in this. He's also correct.
There is a saying that the dose makes the poison. CLA warrants a post or two on its own but it's enough to say for now that there is a toxicity syndrome, reliably induced in rodents, because it's ability to induce lipolysis can be frankly too effective. Including death of adipocytes.
Trans fatty acids are the little brother to CLA as far as lipolysis is concerned.
From the Protons point of view oxidising fats, any fats, will be better than glucose, even with insulin, at inducing reverse electron transport through complex I.
Weight loss, ie fat loss, necessitates the oxidation of lost fat. The better the lipolytic agent, the more fat to oxidise and the more insulin resistance.
Extended fasting is classically a state of profoundly increased fatty acid release from adipocytes and the oxidation of those fatty acids, with insulin resistance being intrinsic to this state. And essential for survival. Protons.
So it is impossible to lose fat without the development of some degree of fat oxidation induced insulin resistance.
CLA is good at lipolysis, trans fats less so but still better than a poke in the eye with a sharp stick.
The thought train which goes on from here is that lipolytic agents should acutely reduce insulin sensitivity directly related to the degree of fat loss. In the long term a lipolytic agent which enforces sustained fat loss will provide the low rate of basal lipolysis intrinsic to small adipocytes and so increase insulin sensitivity, especially if the lipolytic agent is not currently active.
I'm going to talk about clenbuterol next but the other agent of interest is metformin. From the Protons view metformin simply blocks the glycerophosphate shuttle, drops the FADH2 input to the electron transport chain so blunts insulin signalling which needs some degree of ROS generation to happen. Blunting insulin signalling allows lipolysis and suppresses hunger in proportion to these fat loss calories. Once adipocytes are small enough from this blunted insulin signalling we are back in to small adipocytes with low basal lipolysis so increased insulin sensitivity, especially if the metformin has worn off... In humans metformin takes a few weeks to "work". I doubt the degree of fat loss needs to be gross, just enough to reduce basal lipolysis a little.
Back to clenbuterol. Calves this time (at least it's not Bl/6 mice!).
Clenbuterol-Induced Insulin Resistance in Calves Measured by Hyperinsulinemic, Euglycemic Clamp Technique
Basically it's looking at acute treatment with a lipolytic agent. Here are the glucose infusion rates under an hyperinsulinaemic clamp:
Clenbuterol-Induced Insulin Resistance in Calves Measured by Hyperinsulinemic, Euglycemic Clamp Technique
Basically it's looking at acute treatment with a lipolytic agent. Here are the glucose infusion rates under an hyperinsulinaemic clamp:

The black squares are the infusion rates after clenbuterol, the open squares before injection.
It's clear from the bottom graph, while the drug is active, that the treated calves are insulin resistant, requiring significantly less glucose during the hyperinsulinaemic clamp compared to before treatment.
The upper graph shows no effect if you wait 16-25 hours before the clamp, ie until the clenbuterol has worn off. Interestingly the square colours are reversed in this upper graph. Even if the rates are ns different, we still have the calves showing as more insulin sensitive in the aftermath of a period of lipolysis. You can't force lipolysis without shrinking adipocytes. Shrunken adipocytes will always have lower basal lipolysis compared to larger adipocytes. This should show as less insulin resistance. There is a suggestion of that here.
Here are the results tabulated

I was going to go on to talk about chronic clenbuterol and the enhanced insulin sensitivity it provides. Undoubtedly chronic, high dose clenbuterol induces low adipocyte size, muscle hypertrophy and markedly improved insulin sensitivity. But the mechanism becomes complex and convoluted. I spent a little time on this fascinating paper which is comprehensible from the Protons point of view but horribly convoluted by beta receptor down regulation leading to blunted adrenaline signalling. Which affects insulin sensitivity directly.
Clenbuterol prevents epinephrine from antagonizing insulin-stimulated muscle glucose uptake
Clenbuterol prevents epinephrine from antagonizing insulin-stimulated muscle glucose uptake
Fascinating but I'll leave that can of worms alone. It does leave me wondering a little about the acute effects of clenbuterol on fully active beta receptors and their interaction with insulin signalling. Messy. I'll leave the above post unchanged but bear in mind a lot is going on when you take an adrenergic agonist drug, in addition to lipolysis!
Peter
Sunday, November 28, 2021
Are you lino-philic? (2)
Why do Risérus, Willet and Hu get it so wrong? Apart from habit of course. Out by a Ferguson is their usual standard.
Just to regurgitate:
"Taken together, the evidence suggests that replacing saturated fats and trans fatty acids with unsaturated (polyunsaturated and/or monounsaturated) fats has beneficial effects on insulin sensitivity and is likely to reduce risk of type 2 diabetes. Among polyunsaturated fats, linoleic acid from the n-6 series improves insulin sensitivity."
"Taken together, the evidence suggests that replacing saturated fats and trans fatty acids with unsaturated (polyunsaturated and/or monounsaturated) fats has beneficial effects on insulin sensitivity and is likely to reduce risk of type 2 diabetes. Among polyunsaturated fats, linoleic acid from the n-6 series improves insulin sensitivity."
Looking at this study is informative:
Here are the intervention diets

The intervention does exactly what it says on the can. Two five week periods with crossover. The subjects were rock steady for bodyweight throughout the study. Clearly it could not be blinded and the authors speculate that caloric intake might be under reported on the high PUFA arm because there are decades of indoctrination that the PUFA period was "healthy" eating (my phraseology!). I would add that they might even have subconsciously "accidentally" genuinely under eaten rather than under reported. It was only five weeks after all.
Here are the clamp results:

I think it's worth noting that at 120 minutes (Stage of clamp 6) that the glucose infusion rate per unit plasma insulin was still rising in the PUFA period but in the sat fat period the increase had stopped. From the Protons perspective this is the onset of insulin induced insulin resistance, apparently lacking in the PUFA rich period. Not commented on by the researchers but I have the eye of faith. Nice.
Converting the above graph to actual numbers here we have the results table here:

This is all classic Protons.
Protons says insulin signalling makes you fat. Improving insulin sensitivity, ie signalling, will ergo make you fat. Linoleic acid does this better than anything else, pax glitazones. Eventually insulin resistance will occur but only when adipocytes get big enough. This takes longer than five weeks, especially if you succeed in transiently limiting calories to less than those needed to replace calories lost in to adipocytes.
Back to Risérus, Willet and Hu.
To them life appears simple. Skinny people are insulin sensitive. Fat people are insulin resistant. If you could make fat people have the insulin sensitivity of thin people they would become thin. Or at least not diabetic.
Hahahahahahahahahahahaha! Bonk.
They really need Protons.
Peter
Are you trans-phobic? (1)
I've had this paper lying around on my hard drive for some time
Trans-palmitoleic Acid Reduces Adiposity via Increased Lipolysis in a Rodent Model of Diet-Induced Obesity
I don't like it much in terms of writing style, details included/omitted and perspective of the authors but their data look okay and confirm my deepest biases, so I like this aspect.
The usual Bl/6 mice on high fat diet (around 6.5% LA, linoleic acid) vs low fat (around 3.7% LA)
Weights:


You could, from isolated adipocyte studies, make a similar case for elaidic acid (ie shock horror, trans oleic acid, mmmm Crisco).
Replacing Cis Octadecenoic Acid with Trans Isomers in Media Containing Rat Adipocytes Stimulates Lipolysis and Inhibits Glucose utilization

and here are the adiposity index results:

Why do Risérus*, Willet and Hu disagree?
Trans-palmitoleic Acid Reduces Adiposity via Increased Lipolysis in a Rodent Model of Diet-Induced Obesity
I don't like it much in terms of writing style, details included/omitted and perspective of the authors but their data look okay and confirm my deepest biases, so I like this aspect.
The usual Bl/6 mice on high fat diet (around 6.5% LA, linoleic acid) vs low fat (around 3.7% LA)
Weights:
We can see that 4% of calories as trans palmitoleate partially offsets the obesogenic effect of just over 6% linoleic acid in Bl/6 mice.
In terms of adipocyte size the mean surface area on a histology section is normalised:
I would expect normal sized adipocytes to have normal basal lipolysis and not be causing excess FFA release in the presence of insulin. The study didn't look at insulin or insulin resistance but they mention various papers in passing where there are suggestions of this being the case.
You could, from isolated adipocyte studies, make a similar case for elaidic acid (ie shock horror, trans oleic acid, mmmm Crisco).
Replacing Cis Octadecenoic Acid with Trans Isomers in Media Containing Rat Adipocytes Stimulates Lipolysis and Inhibits Glucose utilization
"Compared with oleic acid, both trans isomers reduced (P < 0.01) the amount of glucose converted to cell lipid in both experiments. Glucose oxidation to carbon dioxide also was lower for both trans fatty acids in Experiments 1 (P < 0.05) and 2 (P < 0.06). Lipolytic rates were increased (P < 0.01) in both experiments by replacing oleic acid with either of the trans isomers."
"...and decreased adipocyte size (−44%) versus control rats."
You can make the same case for trans vaccenic acid using whole rats:
Diets enriched in trans-11 vaccenic acid alleviate ectopic lipid accumulation in a rat model of NAFLD and metabolic syndrome
Diets enriched in trans-11 vaccenic acid alleviate ectopic lipid accumulation in a rat model of NAFLD and metabolic syndrome
"...and decreased adipocyte size (−44%) versus control rats."
also worth noting
"[trans vaccenic acid] supplementation also increased metabolic rate (7%)"
Trans vaccenic acid stops the development of metabolic syndrome just as trans palmitoleic acid does. For those of us who consider metabolic syndrome to be the replacement of insulin/sympathic system controlled lipolysis by elevated, uncontrolled, adipocyte size determined basal lipolysis this is exactly what you might expect.
In this next study they replaced 7.2% trans vaccenic acid and 3.4% elaidic acid (original Primex) with palmitate (Primex-Z) while maintaining 24% of the fat as LA. Much as I love palmitic acid it is not an active lipolytic agent in the way that the trans fats are.
Chronic ingestion of Primex-Z, compared with other common fat sources, drives worse liver injury and enhanced susceptibility to bacterial infections
Here are the weights at 16 and 30 weeks, first column is the control Bl/6 mice, second is the trans fat mice, third is the unopposed LA:
In this next study they replaced 7.2% trans vaccenic acid and 3.4% elaidic acid (original Primex) with palmitate (Primex-Z) while maintaining 24% of the fat as LA. Much as I love palmitic acid it is not an active lipolytic agent in the way that the trans fats are.
Chronic ingestion of Primex-Z, compared with other common fat sources, drives worse liver injury and enhanced susceptibility to bacterial infections
Here are the weights at 16 and 30 weeks, first column is the control Bl/6 mice, second is the trans fat mice, third is the unopposed LA:

and here are the adiposity index results:

Sadly again no assessment of insulin function was made but with comparable adiposity to the control mice I wouldn't expect them to be insulin resistant. The corn oil group will have been pushing uncoupling levels of LA. By 30 weeks everything looks pretty much as I would expect it to.
So I might claim not to be trans-phobic. Except I support JK Rowling.
Why do Risérus*, Willet and Hu disagree?
[* That's Risérus as in The Muffin Study and Willet and Hu are the Usual Suspects]
Dietary fats and prevention of type 2 diabetes
"Taken together, the evidence suggests that replacing saturated fats and trans fatty acids with unsaturated (polyunsaturated and/or monounsaturated) fats has beneficial effects on insulin sensitivity and is likely to reduce risk of type 2 diabetes. Among polyunsaturated fats, linoleic acid from the n-6 series improves insulin sensitivity."
Which is, of course, absolute, total bollocks.
"Taken together, the evidence suggests that replacing saturated fats and trans fatty acids with unsaturated (polyunsaturated and/or monounsaturated) fats has beneficial effects on insulin sensitivity and is likely to reduce risk of type 2 diabetes. Among polyunsaturated fats, linoleic acid from the n-6 series improves insulin sensitivity."
Which is, of course, absolute, total bollocks.
There is a reason they make this mistake but this post is too long and rambling already.
Peter
Monday, November 15, 2021
Is vaccine efficacy a statistical illusion?
Just a twitter-ish one liner:
After a chat with Raphi on twitter this might make it clearer. Campaign starts at day one. No results are collected for a week 'cos that's how long it takes. No one know exactly when a given person died because mortality stats are like that and this is not a controlled study situation we're talking about.
Insight delivered on a plate. A clear explanation of the John Dee's Almanac concept. Look how the sizes of populations shift with time on a fixed death rate giving the illusion of efficacy. And also of apparent waning efficacy with time. So elegant, so neat, love it.
Peter
Addendum if it helps:
EDIT Just to clarify, there is no need for the "vaccine" to do anything, you can even assume it's a placebo injection. The effect still occurs. END EDIT
The numbers of deaths collected a week after the campaign started are attributed to week two because that's when they are recorded. This is the source of the error.
If 15 people a day die during week one but are recorded as week two they will be put in to incorrect population sizes because the vaccinated population is rising rapidly and the unvaccinated population size is falling rapidly. A week is a long time in a vaccine roll out.
So the small number of deaths in the initially tiny vaccinated group of week one will be attributed to the significantly larger vaccinated group found in week two. Very few deaths from a very small population are now spread out over a now larger population.
The much larger number of deaths from the much bigger unvaccinated population of week one will be attributed to the now smaller unvaccinated population of week two. The population is smaller because vaccines have been given, which rapidly reduces the size of the unvaccinated population.
In the vaccinated group too small a number of deaths is spread through too large a number of people, hence a low incidence/person days. Vaccine appears to work.
In the unvaccinated too many deaths (it was a very big group in week one) are attributed to a population reduced by the number who have been vaccinated by the rollout. So a much higher figure per person days is found.
Don't start me on how this makes being unvaccinated intrinsically dangerous and how the 'rona vacc appears to protect agains all cause mortality. Just more artifact.
The graphs come out as in the linked blog post.
The need for graphing mortality curves by date of death vs date of reporting is well known from plotting peaks of waves from peaks of deaths. If a study uses date reported rather than date of occurrence, it's possibly junk. It can take months to get death numbers by date of occurrence vs reported in the real world. Some mortality data from the UK ONS will be delayed by the time needed for a coroner's inquest.
Peter
Thursday, November 04, 2021
!Kung Bushmen and mongongo nuts again
Back in the mongongo nuts post I suggested that conjugated linoleic acid (CLA), which shows the hallmarks of a lipolytic agent, derived from the alpha eleostearic acid in the nuts might offset the obesogenic effect of the common or garden linoleic acid (LA) present in roughly equal amounts.
Metabolic Responses to Oral Glucose in the Kalahari Bushmen
"Since an overnight fast would probably have been broken (owing to the almost continuous eating pattern of the Bushmen when food is available), we performed tests in the afternoon, after four hours of observed rest and fasting."
What does an oral glucose tolerance test look like in a !Kung bushman?


The closed circles are perfusion with LA. So perhaps it's not too surprising that the !Kung are hypoinsulinaemic. And it doesn't matter because they are also very insulin sensitive. This balances out.
We have the !Kung consuming anything from zero to 1000kcal/d of mongongo nuts per day, average 800kcal/d, ie about 40% of calories:
You can download the full text from Scihub. It's a nice read.
So. If this is the case you have a pair of opposing effects, from the Protons point of view the LA is insulin sensitising and will allow excess insulin signalling to distend adipocytes when they should be signalling that they are full. Under fasting it can allow relative hypoglycaemia, encouraging food intake, but that's not a feature of this study. All subjects were only fasted for four hours.
At exactly the same time the alpha eleostearic acid derived CLA will be facilitating the release of FFAs from adipocytes which means that fat cells stay approximately the correct size and those FFAs are available to be perceived by the brainstem. So, from the adipocyte point of view we have excess calories-in and excess calories-out concurrently. If the adipocytes never distend we will never have to deal with size-derived excess basal lipolysis and the associated appropriate insulin resistance.
Now we can look at how that might explain the observation in this paper
Metabolic Responses to Oral Glucose in the Kalahari Bushmen
"Since an overnight fast would probably have been broken (owing to the almost continuous eating pattern of the Bushmen when food is available), we performed tests in the afternoon, after four hours of observed rest and fasting."
The Bushmen eat very frequently when food is available but never enough in total to become obese.
What does an oral glucose tolerance test look like in a !Kung bushman?
Like this:

As the authors comment
"Mean glucose levels were higher in the Bushmen at all stages, with significant differences at 0 and 120 minutes. Indeed, by lax criteria of evaluation (Jackson et al., 1970), their mean two-hour post-glucose level of 121 mg/ 100 ml could be regarded as falling within the "diabetic" range. Conversely, the Bushmen exhibited insulinopenia throughout the test, and this was significant at 0 and 60 minutes."
"Mean glucose levels were higher in the Bushmen at all stages, with significant differences at 0 and 120 minutes. Indeed, by lax criteria of evaluation (Jackson et al., 1970), their mean two-hour post-glucose level of 121 mg/ 100 ml could be regarded as falling within the "diabetic" range. Conversely, the Bushmen exhibited insulinopenia throughout the test, and this was significant at 0 and 60 minutes."
These people are insulin sensitive, as you would expect from a high LA intake. However they don't become obese because they never secrete very much insulin, ignoring the CLA. Does anyone recall this image of an isolated, perfused rat pancreas?

The closed circles are perfusion with LA. So perhaps it's not too surprising that the !Kung are hypoinsulinaemic. And it doesn't matter because they are also very insulin sensitive. This balances out.
In some ways they remind me of Jim Johnson's reduced insulin gene dosed mice in which, during early life, some glucose intolerance was present secondary to hypoinsulinaemia but this self corrected with age. Clearly the !Kung, with normal insulin genetics, are quite capable of becoming obese on an high LA diet simply by ramping up their insulin secretion in response to a mixed diet and it is the CLA which stops this. So they never develop the problems secondary to distended adipocytes.
Other explanations welcome.
What would have been really interesting would have been an insulin tolerance test which looks at insulin sensitivity without needing any confounding contribution from (decreased) pancreatic insulin secretion. I think we can assume that there would have been a profound fall in blood glucose in response to exogenous insulin.
Peter
Tuesday, November 02, 2021
Are COVID-19 vaccines useful? (3)
Here is an interesting twitter thread by a mainstream author in which he discusses the problems in the Swedish observational study preprint
Effectiveness of Covid-19 Vaccination Against Risk of Symptomatic Infection, Hospitalization, and Death Up to 9 Months: A Swedish Total-Population Cohort Study
where vaccine efficacy drops to zero by 200 days and becomes negative there-after.

From https://t.me/JohnDeesAlmanac/639
Consider with caution. There appear to be lots of ways of looking at lots of data!
where vaccine efficacy drops to zero by 200 days and becomes negative there-after.
Life is probably not that simple and natural infection means that by the 200 day mark you are comparing recovered field infected people with naive vaccinated people, at least I think that's his argument. So apples are being compared to progressively ripening oranges.
What’s more interesting is a related second twitter thread of his in which he discusses this study of the fully vaccinated only (ie more than 14 days post second dose, mostly)
Immune Responses in Fully Vaccinated Individuals Following Breakthrough Infection with the SARS-CoV-2 Delta Variant in Provincetown, Massachusetts
which looks at the short term consequences of vaccine failure (“breakthrough” infections) on a number of parameters. The vaccinated study population were jabbed in the near absence of circulating virus so the normal 14 days of immunosuppression didn’t cause an infection problem. Later on a wave of field infection passed through the area as the virus "virused" in its normal manner, though somewhat out of season.
Immune Responses in Fully Vaccinated Individuals Following Breakthrough Infection with the SARS-CoV-2 Delta Variant in Provincetown, Massachusetts
which looks at the short term consequences of vaccine failure (“breakthrough” infections) on a number of parameters. The vaccinated study population were jabbed in the near absence of circulating virus so the normal 14 days of immunosuppression didn’t cause an infection problem. Later on a wave of field infection passed through the area as the virus "virused" in its normal manner, though somewhat out of season.
Here’s the most important figure, it's as close as the study got to asking useful questions:

Section A simply tells us that having a vaccine systemic anti-spike antibody titre of around 1 in 200-ish derived from the vaccine does nothing to protect against infection and that field virus exposure bumps this up to 1 in 2,000-ish in recovery. Shrug, we should expect that there would be a marked response to the original antigen seen by the immune system, especially in the systemic circulation.
What is much more interesting is section B. This shows systemic IgG response to nucleocapsid antigen, which as essentially zero in people who were vaccinated but not infected, on the left side of the graph. The right hand side, filled circles, shows people who were vaccinated and then became infected. There is an anti-nucleocapsid systemic IgG response of a magnitude comparable to the anti-spike IgG response produced by the initial vaccination. This suggest that OAS is present but does not eliminate the response to other antigens of the field virus.
Now, whether a spike-only antibody titre of 1:200 is protective against anything is an open point, so whether a broad antigen antibody response, illustrated by anti nucleocapsid response, is effective against anything either is also a somewhat open point. All of the infected people were survivors and all had mild illness. It seems like a religious question as to whether mild disease was due to huge anti-spike response or modest multi-epitope response. Or catching the virus in July in northern latitudes.
Ultimately the vast majority (but clearly not all) of people are going to survive exposure to SARS-CoV-2, especially if it happens in mid summer. The vaccine appears to allow field infection to produce a broad antigen, probably sterilising immunity. OAS still shows as the marked anti-spike response to field virus but this does not stop recovery or a more general immune response.
And because the vaccine does nothing to eliminate spread we will eventually have enough people infected to then limit transmission to endemic levels.
Peter
Controversial addendum: This link is to a less-than-preprint conversational musing piece from someone who has access to NHS data of a detail beyond anyone's wildest dreams in Twitterland, hence it's on Telegraph. So some serious caveats have to be applied but his conclusion is that the vaccines do absolutely nothing. At all. An interesting idea.
From https://t.me/JohnDeesAlmanac/639
Consider with caution. There appear to be lots of ways of looking at lots of data!
Sunday, October 24, 2021
Are COVID-19 vaccines useful? (2)
I really, really don't like this:
Nasal prevention of SARS-CoV-2 infection by intranasal influenza-based boost vaccination
I was very pleased to receive the OAS source paper from Mike Eades (thanks Mike!) and it's a great read. An old paper from 1960, written in the style of the time, giving a basic idea discussed in almost conversational terms by a single author, with enough data to back up the idea, explaining where it came from. I can't see the original paper on tinternet although there are lots citing it:
ON THE DOCTRINE OF ORIGINAL ANTIGENIC SIN
Nasal prevention of SARS-CoV-2 infection by intranasal influenza-based boost vaccination
I picked it up via a tweet from Gabor Erdosi in the aftermath of the excellent discussion he had with Raphi, available on Youtube.
The only fundamental problem I have with their discussion was about original antigenic sin and the above pertains directly to this.
I was very pleased to receive the OAS source paper from Mike Eades (thanks Mike!) and it's a great read. An old paper from 1960, written in the style of the time, giving a basic idea discussed in almost conversational terms by a single author, with enough data to back up the idea, explaining where it came from. I can't see the original paper on tinternet although there are lots citing it:
ON THE DOCTRINE OF ORIGINAL ANTIGENIC SIN
EDIT location for the first page provided via eugyppius END EDIT
NEXT EDIT Full text here, thanks to Raphi END EDIT AGAIN
So. My fundamental difference in viewpoint to Gabor is that my expectation is that OAS from the mRNA vaccines would be limited to the systemic immune system and the respiratory mucosal immune system would be free from OAS and so able to mount a broad, effective response to produce sterilising immunity to a field infection. Whatever the evolution of spike protein to antibody avoidance, the respiratory mucosal system should stay clean.
So. My fundamental difference in viewpoint to Gabor is that my expectation is that OAS from the mRNA vaccines would be limited to the systemic immune system and the respiratory mucosal immune system would be free from OAS and so able to mount a broad, effective response to produce sterilising immunity to a field infection. Whatever the evolution of spike protein to antibody avoidance, the respiratory mucosal system should stay clean.
The above technique of giving an intranasal live attenuated influenza vaccine at the same time as a systemic IgG inducing mRNA based vaccine appears to trigger respiratory mucosal IgA formation to the systemic spike protein antigen. You "kick" the respiratory system with an attenuated influenza vaccine and the "awake" respiratory immune system "notices" and responds to what should have been a systemic-only spike protein stimulus.
In the UK we already have routine childhood intranasal influenza vaccines. I'm not anti-vax, my kids get the intranasal flu vaccine, FWIW. Assuming mRNA Covid vaccines are made mandatory for children (peak stupidity, but nothing surprises me) I can see that the logistics of delivering both vaccines on the same day might favour doing both at once.
So for kids OAS would be effectively extended from systemic IgG to mucosal IgA. Which might well blunt a correct, broad antigenic response within the airway at the time of a subsequent field virus infection.
This sounds like a very, very, very stupid thing to do.
So it will probably become standard practice, even mandatory.
Peter
On the plus side the mucosal IgA might be sterilising, if anti-spike antibodies are enough to kill all of the virus. This will limit time available for selecting an anti-spike antibody evading strain of virus. But the pressure will be still there.
It's like giving a full therapeutic dose of methicillin to a patient with a methicillin susceptible staphylococcal infection. It works. You have to do it. You have to kill the staph completely. But one day you will still successfully select for MRSA... It will happen. It already has.
Friday, October 22, 2021
Are COVID-19 vaccines useful?
Mike Eades updated the current copy of the Arrow to include Alex Berenson's observation that the COVID-19 vaccine surveillance report Week 42 from the UK Health Security Agency (ie the UK Government, such as it is) details that the phenomenon of Original Antigenic Sin is clearly being demonstrated in the UK covid antibody data.
This concept is very simple and predicts that if you are exposed to a single antigen (here the spike protein derived from an mRNA vaccine) your immune system will prioritise a response to that single antigen in preference to other antigens when presented with a mixed antigen soup, as in the whole virus during a subsequent field infection.
So, in double vaccinated individuals you have preferential response to the spike protein over nucleocapsid protein as assessed by antibody titres. Page 23 if you want to have a look:
"recent observations from UK Health Security Agency (UKHSA) surveillance data that N [nucleocapsid] antibody levels appear to be lower in individuals who acquire infection following 2 doses of vaccination."
ie the vaccine screws your immune response to nucleocapsid.
Somewhat.
However the UKHSA only monitor anti-spike protein and anti-nucleocapsid antibodies because these allow us to distinguish between vaccine exposure and field infection. Obviously field infection triggers many more immune responses in addition to those against spike and nucleocapsid proteins, none of which need to be monitored to get this information.
As we vaccinate using the spike protein alone we will actively favour the survival of vaccine evading mutations. Boosters will speed this up.
So, are we all going to die?
I think not. UKHSA is monitoring antibodies. These are being surveyed in recovered patients.
"Recovered" is the word.
Ultimately triple (and greater, eventually) vaccinated people, so long as the vaccine is spike protein based, will eventually end up behaving as though they are unable to even "see" the spike protein, their anti-spike antibodies will be present but will do nothing. Spike evasion will have happened and the selection pressure will no longer be present. Lots of anti-spike antibodies, no interaction with the spike, no further selection pressure.
Vaccinated people will have to run on non-spike immune response, which will still be broad and still work. It may not be as effective as in the non vaccinated, because the immune system prioritises large amounts of useless spike response, but most people will still survive (unless they have chosen to be a poorly controlled diabetic, diagnosed or in-situ of course) as they are doing currently.
In some ways I can see some use for the vaccine and the idea of vaccine passports.
Aside: Of course using vaccine passports for anything, especially to pauperise and exclude the unvaccinated, will come with a sh!tload of human rights violations in addition to the health problems automatically generated by pauperisation per se. This is morally reprehensible and unforgivable. It's happening now if you live in the wrong country. Don't you love politicians? End aside.
At the start of the pandemic certain groups of people were thrown under the bus as regards covid. These are people who do actual work. Supermarket checkout cashiers, bus drivers, garbage collectors, postal workers, truck drivers, construction workers. Others, like myself, were given several months leave on 80% of salary with a big garden during some of the sunniest Spring weather I can recall. So we "let the virus rip" through people who actually do jobs ("essential workers") and paid loafers like myself ££££ of my children's and probably grandchildren's money to stay at home and "avoid" the virus. For a while.
Now the vaccine is here and the virus is in reality being allowed to rip through the rest of society, including the laptop classes. Clearly vaccine passports will actively concentrate vaccinated people in to crowded places and so maximise transmission. The UKHSE report cited above also reports vaccinated people are a lot better at getting infected compared to the unvaccinated, interestingly enough. Provided these people do survive (and most will) then they will end up with a ton of useless anti-spike "immunity" plus enough real immunity to other components of the virus to survive future exposure to that virus. We need this.
That should be enough.
Peter
PS I can live without the human rights violations which seem to be endemic at the moment. Or should I say epidemic or pandemic???
Subscribe to:
Posts (Atom)
